Cosentyx® (secukinumab) [1]
1. ИМЕ НА ЛЕКАРСТВЕНИЯ ПРОДУКТ
Cosentyx 150 mg инжекционен разтвор в предварително напълнена спринцовка
Cosentyx 300 mg инжекционен разтвор в предварително напълнена спринцовка
Cosentyx 150 mg инжекционен разтвор в предварително напълнена писалка
Cosentyx 300 mg инжекционен разтвор в предварително напълнена писалка
2. КАЧЕСТВЕН И КОЛИЧЕСТВЕН СЪСТАВ
Cosentyx 150 mg инжекционен разтвор в предварително напълнена спринцовка
Всяка предварително напълнена спринцовка съдържа 150 mg секукинумаб (secukinumab) в 1 ml.
Cosentyx 300 mg инжекционен разтвор в предварително напълнена спринцовка
Всяка предварително напълнена спринцовка съдържа 300 mg секукинумаб (secukinumab) в 2 ml.
Cosentyx 150 mg инжекционен разтвор в предварително напълнена писалка
Всяка предварително напълнена писалка съдържа 150 mg секукинумаб (secukinumab) в 1 ml
Cosentyx 300 mg инжекционен разтвор в предварително напълнена писалка
Всяка предварително напълнена писалка съдържа 300 mg секукинумаб (secukinumab) в 2 ml.
Секукинумаб е рекомбинантно изцяло човешко моноклонално антитяло, получено от клетки от яйчник на китайски хамстер (CHO).
За пълния списък на помощните вещества вижте точка 6.1.
3. ЛЕКАРСТВЕНА ФОРМА
Инжекционен разтвор (инжекция)
Разтворът е бистър и безцветен до бледожълт.
4. КЛИНИЧНИ ДАННИ
4.1 Терапевтични показания
Плакатен псориазис при възрастни
Cosentyx е показан за лечение на умерено тежък до тежък плакатен псориазис при възрастни, които са кандидати за системна терапия.
Плакатен псориазис при педиатрични пациенти
Cosentyx е показан за лечение на умерен до тежък плакатен псориазис при деца и юноши на възраст над 6 години, които са кандидати за системна терапия.
Псориатичен артрит
Cosentyx, самостоятелно или в комбинация с метотрексат (MTX), е показан за лечение на активен псориатичен артрит при възрастни пациенти, при които предшестващата терапия с модифициращи болестта антиревматоидни средства (DMARD) е била недостатъчна (вж. точка 5.1).
Аксиален спондилоартрит (axSpA)
Анкилозиращ спондилит (AS, рентгенографски потвърден аксиален спондилоартрит)
Cosentyx е показан за лечение на активен анкилозиращ спондилит при възрастни, които не са се повлияли достатъчно от конвенционалната терапия.
Нерентгенографски потвърден аксиален спондилоартрит (nr-axSpA)
Cosentyx е показан за лечение на активен нерентгенографски потвърден аксиален спондилоартрит с обективни признаци на възпаление, които включват повишен С-реактивен протеин (CRP) и/или данни от ядрено-магнитен резонанс (ЯМР) при възрастни, които са имали недостатъчен отговор към лечение с нестероидни противовъзпалителни средства (НСПВС).
Ювенилен идиопатичен артрит (ЮИА)
Артрит, свързан с ентезит (еnthesitis-related arthritis, ERA)
Cosentyx, самостоятелно или в комбинация с метотрексат (MTX), е показан за лечение на активен артрит, свързан с ентезит, при пациенти на възраст на и над 6 години, при които заболяването има недостатъчно повлияване, или имат непоносимост, към конвенционална терапия (вж. точка 5.1).
Ювенилен псориатичен артрит (ЮПсА)
Cosentyx, самостоятелно или в комбинация с метотрексат (MTX), е показан за лечение на активен ювенилен псориатичен артрит при пациенти на възраст на и над 6 години, при които заболяването има недостатъчно повлияване, или имат непоносимост, към конвенционална терапия (вж. точка 5.1).
4.2 Дозировка и начин на приложение
Cosentyx е показан за приложение под ръководството и контрола на лекар с опит в диагностицирането и лечението на заболяванията, за които Cosentyx е показан.
Дозировка
Плакатен псориазис при възрастни
Препоръчителната доза е 300 mg секукинумаб, приложени чрез подкожна инжекция, като първоначално се прилага на седмица 0, 1, 2, 3 и 4, а впоследствие като ежемесечна поддържаща доза. Въз основа на клиничния отговор, допълнителна полза при пациенти с телесно тегло 90 kg или повече се постига с поддържаща доза 300 mg на всеки 2 седмици. Всяка доза от 300 mg се прилага като една подкожна инжекция от 300 mg или като две подкожни инжекции от 150 mg.
Плакатен псориазис при педиатрични пациенти (юноши и деца на възраст над 6 години)
Препоръчителната доза е въз основа на телесното тегло (Таблица 1), приложена чрез подкожна инжекция, като първоначално се прилага на седмица 0, 1, 2, 3 и 4, а впоследствие като ежемесечна поддържаща доза. Всяка доза от 75 mg се прилага като една подкожна инжекция от 75 mg. Всяка доза от 150 mg се прилага като една подкожна инжекция от 150 mg. Всяка доза от 300 mg се прилага като една подкожна инжекция от 300 mg или като две подкожни инжекции от 150 mg.
Таблица 1 Препоръчителна доза за плакатен псориазис при педиатрични пациенти
|
Телесно тегло към момента на прилагане |
Препоръчителна доза |
|
<25 kg |
75 mg |
|
25 до <50 kg |
75 mg |
|
≥50 kg |
150 mg (*може да се увеличи до 300 mg) |
*Някои пациенти може да получат допълнителна полза от по-високата доза.
150 mg и 300 mg инжекционен разтвор в предварително напълнена спринцовка и предварително напълнена писалка не е показан за приложение при педиатрични пациенти с телесно тегло <50 kg. Cosentyx може да се предлага в други концентрации и/или лекарствени форми в зависимост от индивидуалните нужди при лечението.
Псориатичен артрит
При пациенти със съпътстващ умерено тежък до тежък плакатен псориазис, вижте препоръката за плакатен псориазис при възрастни.
При пациенти, които не са се повлияли достатъчно от проведената анти-TNFα терапия, препоръчителната доза е 300 mg, приложени чрез подкожна инжекция, като първоначално се прилага на седмица 0, 1, 2, 3 и 4, а впоследствие като ежемесечна поддържаща доза. Всяка доза от 300 mg се прилага като една подкожна инжекция от 300 mg или като две подкожни инжекции от 150 mg.
При другите пациенти препоръчителната доза е 150 mg, приложени чрез подкожна инжекция, като първоначално се прилага на седмица 0, 1, 2, 3 и 4, а впоследствие като ежемесечна поддържаща доза. Въз основа на клиничния отговор дозата може да се повиши до 300 mg.
Аксиален спондилоартрит (аxial spondyloarthritis, axSpA)
Анкилозиращ спондилит (AS, рентгенографски потвърден аксиален спондилоартрит)
Препоръчителната доза е 150 mg, приложени чрез подкожна инжекция, като първоначално се прилага на седмица 0, 1, 2, 3 и 4, а впоследствие като ежемесечна поддържаща доза. Въз основа на клиничния отговор, дозата може да се повиши до 300 mg. Всяка доза от 300 mg се прилага като една подкожна инжекция от 300 mg или като две подкожни инжекции от 150 mg.
Нерентгенографски потвърден аксиален спондилоартрит (non-radiographic axial spondyloarthritis, nr-axSpA)
Препоръчителната доза е 150 mg, приложени чрез подкожна инжекция, като първоначално се прилага на седмица 0, 1, 2, 3 и 4, а впоследствие като ежемесечна поддържаща доза.
Ювенилен идиопатичен артрит (ЮИА)
Артрит, свързан с ентезит (ERA) и ювенилен псориатичен артрит (ЮПсА)
Препоръчителната доза е въз основа на телесното тегло (Таблица 2) и се прилага чрез подкожна инжекция на седмица 0, 1, 2, 3 и 4, а впоследствие като ежемесечна поддържаща доза. Всяка доза от 75 mg се прилага като една подкожна инжекция от 75 mg. Всяка доза от 150 mg се прилага като една подкожна инжекция от 150 mg.
Таблица 2 Препоръчителна доза за ювенилен идиопатичен артрит
|
Телесно тегло към момента на прилагане |
Препоръчителна доза |
|
<50 kg |
75 mg |
|
≥50 kg |
150 mg |
150 mg и 300 mg инжекционен разтвор в предварително напълнена спринцовка и предварително напълнена писалка не е показан за приложение при педиатрични пациенти с телесно тегло <50 kg. Cosentyx може да се предлага в други концентрации и/или лекарствени форми в зависимост от индивидуалните нужди при лечението.
При всички показания, изброени по-горе, наличните данни показват, че клиничен отговор обикновено се постига в рамките на 16 седмици лечение. Необходимо е да се обмисли преустановяване на лечението при пациенти, които не показват повлияване до 16-та седмица от лечението. Някои пациенти, които първоначално имат частично повлияване, е възможно впоследствие да се подобрят, при продължаване на лечението след 16-та седмица.
Специални популации
Пациенти в старческа възраст (на и над 65 години)
Не е необходимо коригиране на дозата (вж. точка 5.2).
Бъбречно увреждане/чернодробно увреждане
Cosentyx не е проучван в тези пациентски популации. Не могат да бъдат дадени препоръки относно дозата.
Педиатрична популация
Безопасността и ефикасността на Cosentyx при деца с плакатен псориазис и в категориите на ювенилен идиопатичен артрит (ЮИА) ERA и ЮПсА на възраст под 6 години не са установени.
Безопасността и ефикасността на Cosentyx при деца на възраст под 18 години при други показания все още не са установени. Липсват данни.
Начин на приложение
Cosentyx се прилага чрез подкожна инжекция. Ако е възможно, областите по кожата, засегнати от псориазис трябва да се избягват като места на инжектиране. Да не се разклаща спринцовката или писалката.
След подходящо обучение в техниката на поставяне на подкожна инжекция, пациентите могат сами да си инжектират Cosentyx или инжекцията да се приложи от болногледач, ако лекарят прецени, че това е уместно. Лекарят обаче трябва да осигури съответното проследяване на пациентите. Пациентите или болногледачите трябва да бъдат инструктирани да инжектират цялото количество Cosentyx, съгласно указанията, описани в листовката. Подробни указания относно начина на приложение са дадени в листовката.
4.3 Противопоказания
Свръхчувствителност към активното(ите) вещество(а) или към някое от помощните вещества, изброени в точка 6.1.
Клинично значими активни инфекции, напр. активна туберкулоза (вж. точка 4.4).
4.4 Специални предупреждения и предпазни мерки при употреба
Проследимост
За да се подобри проследимостта на биологичните лекарствени продукти, името и партидният номер на приложения продукт трябва ясно да се записват.
Инфекции
Секукинумаб има потенциала да повишава риска от инфекции. По време на постмаркетинговия период са наблюдавани сериозни инфекции при пациенти, получаващи секукинумаб. Необходимо е повишено внимание, когато се обмисля приложението на секукинумаб при пациенти с хронична инфекция или анамнеза за рецидивиращи инфекции.
Пациентите трябва да бъдат инструктирани да потърсят медицинска помощ, ако се появят признаци или симптоми, предполагащи наличието на инфекция. Ако някой пациент развие сериозна инфекция, той трябва внимателно да се наблюдава и секукинумаб не трябва да се прилага, докато инфекцията не отзвучи.
В клиничните проучвания са наблюдавани инфекции при пациентите, получаващи секукинумаб (вж. точка 4.8). Повечето от тях са били леки до умерени по тежест инфекции на горни дихателни пътища, като назофарингит, и не са изисквали преустановяване на лечението.
Във връзка с механизма на действие на секукинумаб, съобщенията за несериозни кожнолигавични инфекции с кандида, са по-чести при секукинумаб, отколкото при плацебо, по време на клиничните проучвания при псориазис (3,55 на 100 пациентогодини за секукинумаб 300 mg спрямо 1,00 на 100 пациентогодини за плацебо) (вж. точка 4.8).
От клиничните проучвания не се съобщава за повишена предразположеност към туберкулоза. Независимо от това, секукинумаб не трябва да се прилага при пациенти с активна туберкулоза. Трябва да се обмисли провеждането на противотуберкулозна терапия преди започване на лечението със секукинумаб при пациентите с латентна туберкулоза.
Възпалителни чревни заболявания (включително болест на Crohn и улцерозен колит)
Има съобщения за нови случаи или за случаи на обостряне на възпалителни чревни заболявания при употребата на секукинумаб (вж. точка 4.8). Секукинумаб не се препоръчва при пациенти с възпалително чревно заболяване. Ако пациент развие признаци и симптоми на възпалително чревно заболяване или получи обостряне на вече съществуващо възпалително чревно заболяване, употребата на секукинумаб трябва да се преустанови и да се започне подходящо лечение.
Реакции на свръхчувствителност
В клиничните проучвания са наблюдавани редки случаи на анафилактични реакции при пациенти, лекувани със секукинумаб. Ако се появи анафилактична реакция или други реакции на свръхчувствителност, приложението на секукинумаб трябва незабавно да се спре и да се започне съответната терапия.
Индивиди с чувствителност към латекс – само за Cosentyx 150 mg инжекционен разтвор в предварително напълнена спринцовка и 150 mg инжекционен разтвор в предварително напълнена писалка
Подвижната капачка на иглата на Cosentyx 150 mg инжекционен разтвор в предварително напълнена спринцовка и Cosentyx 150 mg инжекционен разтвор в предварително напълнена писалка съдържа производно на естествения каучук. До момента не е установено наличие на естествен каучук в подвижната капачка. Въпреки че употребата на Cosentyx 150 mg инжекционен разтвор в предварително напълнена спринцовка и Cosentyx 150 mg инжекционен разтвор в предварително напълнена писалка при индивиди с чувствителност към латекс не е проучвана, съществуването на потенциален риск от развитие на реакции на свръхчувствителност не може напълно да се изключи.
Ваксини
Не трябва да се прилагат живи ваксини едновременно със секукинумаб.
При пациентите, получаващи секукинумаб могат да се прилагат инактивирани или убити ваксини. В едно проучване, след прилагане на менингококова ваксина и инактивирана ваксина срещу грип, сходен процент здрави доброволци, третирани със секукинумаб 150 mg и такива, третирани с плацебо, са били в състояние да изградят адекватен имунен отговор от поне 4-кратно повишаване на титъра на антителата срещу приложените ваксини срещу менингококи и грип. Данните предполагат, че секукинумаб не потиска хуморалния имунен отговор към ваксините срещу менингококи и грип.
Преди започване на лечение с Cosentyx се препоръчва педиатричните пациенти да получат всички подходящи за възрастта ваксини, според актуалните препоръки за имунизация.
Съпътстваща имуносупресивна терапия
В проучванията при псориазис безопасността и ефикасността на секукинумаб в комбинация с имуносупресори, включително биологични лекарствени продукти или фототерапия, не са оценявани. Секукинумаб е прилаган съпътстващо с метотрексат (MTX), сулфасалазин и/или кортикостероиди в проучвания при артрит (включително при пациенти с псориатичен артрит и анкилозиращ спондилит). Необходимо е повишено внимание, когато се обмисля съпътстващо използване на други имуносупресори и секукинумаб (вж. също точка 4.5).
4.5 Взаимодействие с други лекарствени продукти и други форми на взаимодействие
Не трябва да се прилагат живи ваксини едновременно със секукинумаб (вж. също точка 4.4).
В проучване при възрастни пациенти с плакатен псориазис не са наблюдавани взаимодействия между секукинумаб и мидазолам (субстрат на CYP3A4).
Не са наблюдавани взаимодействия при прилагане на секукинумаб съпътстващо с метотрексат (MTX) и/или кортикостероиди по време на проучвания при артрит (включително при пациенти с псориатичен артрит и аксиален спондилоартрит).
4.6 Фертилитет, бременност и кърмене
Жени с детероден потенциал
Жените с детероден потенциал трябва да използват ефективни методи на контрацепция по време на лечението и в продължение на поне 20 седмици след лечението.
Бременност
Няма достатъчно данни от употребата на секукинумаб при бременни жени.
Проучванията при животни не показват преки или непреки вредни ефекти, свързани с репродуктивната токсичност (вж. точка 5.3). Като предпазна мярка е за предпочитане да се избягва употребата на Cosentyx по време на бременност.
Кърмене
Не е известно дали секукинумаб се екскретира в кърмата. Имуноглобулините се екскретират в кърмата и не е известно, дали секукинумаб има системна абсорбция след поглъщане. Поради възможността за възникване на нежелани реакции при кърмачетата вследствие употребата на секукинумаб, трябва да се вземе решение, дали да се преустанови кърменето по време на лечението и в продължение на поне 20 седмици след лечението, или да се преустанови терапията с Cosentyx, като се вземат предвид ползата от кърменето за детето и ползата от терапията за жената.
Фертилитет
Ефектът на секукинумаб върху фертилитета при хора не е оценен. Проучванията при животни не показват преки или непреки вредни ефекти по отношение на фертилитета.
4.7 Ефекти върху способността за шофиране и работа с машини
Cosentyx не повлиява или повлиява пренебрежимо способността за шофиране и работа с машини.
4.8 Нежелани лекарствени реакции
Обобщение на профила за безопасност
Най-често съобщаваните нежелани реакции са инфекции на горни дихателни пътища (17,7%) (най-често назофарингит, ринит).
Табличен списък на нежеланите реакции
Нежеланите реакции от клинични проучвания и от постмаркетингови съобщения (Таблица 3) са изброени, съгласно MedDRA, по системо-органни класове. В рамките на всеки системо-органен клас нежеланите реакции са подредени по честота, като най-честите реакции са първи. В рамките на всяко групиране по честота нежеланите реакции са представени в низходящ ред по отношение на тяхната сериозност. В допълнение, съответстващата категория по честота за всяка нежелана реакция е базирана на следната конвенция: много чести (≥1/10); чести (≥1/100 до <1/10); нечести(≥1/1 000 до <1/100); редки (≥1/10 000 до <1/1 000); много редки (<1/10 000); и с неизвестна честота (от наличните данни не може да бъде направена оценка).
Над 18 000 пациенти са лекувани със секукинумаб в заслепени и открити клинични проучвания при различни показания (плакатен псориазис, псориатичен артрит, аксиален спондилоартрит и други автоимунни заболявания), което съответства на експозиция 30 565 пациентогодини. От тях над 11 700 пациенти са били с експозиция на секукинумаб в продължение поне на една година. Профилът на безопасност на секукинумаб е един и същ при всички показания.
Таблица 3 Списък на нежеланите реакции в клиничните проучвания1) и от постмаркетинговия опит
|
Системо-органен клас |
Честота |
Нежелана реакция |
|
Инфекции и инфестации |
Много чести |
Инфекции на горни дихателни пътища |
|
Чести |
Лабиален херпес |
|
|
Тинеа педис |
||
|
Нечести |
Орална кандидоза |
|
|
Отитис екстерна |
||
|
Инфекции на долните дихателни пътища |
||
|
С неизвестна честота |
Лигавична и кожна кандидоза (включително езофагеална кандидоза) |
|
|
Нарушения на кръвта и лимфната система |
Нечести |
Неутропения |
|
Нарушения на имунната система |
Редки |
Анафилактични реакции |
|
Нарушения на нервната система |
Чести |
Главоболие |
|
Нарушения на очите |
Нечести |
Конюнктивит |
|
Респираторни, гръдни и медиастинални нарушения |
Чести |
Ринорея |
|
Стомашно-чревни нарушения |
Чести |
Диария |
|
Чести |
Гадене |
|
|
Нечести |
Възпалително заболяване на червата |
|
|
Нарушения на кожата и подкожната тъкан |
Нечести |
Уртикария |
|
Дисхидротична екзема |
||
|
Редки |
Ексфолиативен дерматит2) |
|
|
Хиперсензитивен васкулит |
||
|
Общи нарушения и ефекти на мястото на приложение |
Чести |
Умора |
|
1) Плацебо-контролирани клинични проучвания (фаза III) при пациенти с плакатен псориазис, псориатичен артрит, анкилозиращ спондилит и нерентгенографски потвърден аксиален спондилоартрит, с експозиция на 300 mg, 150 mg, 75 mg или плацебо, с продължителност на лечението до 12 седмици (псориазис) или до 16 седмици (псориатичен артрит, анкилозиращ спондилит и нерентгенографски потвърден аксиален спондилоартрит). 2) Съобщени са случаи при пациенти с диагноза псориазис. |
||
Описание на избрани нежелани реакции
Инфекции
В плацебо-контролирания период на клиничните проучвания при плакатен псориазис (общо 1 382 пациенти са третирани със секукинумаб и 694 пациенти са третирани с плацебо с продължителност до 12 седмици) инфекции се съобщават при 28,7% от пациентите на лечение със секукинумаб спрямо 18,9% при пациентите на лечение с плацебо. Повечето от инфекциите не са били сериозни и са били леки до умерени по тежест инфекции на горни дихателни пътища, като назофарингит, които не изискват преустановяване на лечението. Наблюдава се повишаване на лигавичната и кожната кандидоза, в съответствие с механизма на действие, но случаите са били леки до умерени по тежест, не са били сериозни, повлияли са се от стандартно лечение и не са изисквали преустановяване на лечението. Сериозни инфекции възникват при 0,14% от пациентите на лечение със секукинумаб и при 0,3% от пациентите на плацебо (вж. точка 4.4).
В рамките на целия период на лечение (общо 3 430 пациенти, третирани със секукинумаб в продължение до 52 седмици за повечето пациенти), инфекции се съобщават при 47,5% от пациентите на лечение със секукинумаб (0,9 на пациентогодина при проследяване). Сериозни инфекции се съобщават при 1,2% от пациентите на лечение със секукинумаб (0,015 на пациентогодина при проследяване).
Честотата на инфекциите, наблюдавана в клиничните проучвания при псориатичен артрит и аксиален спондилоартрит (анкилозиращ спондилит и нерентгенографски потвърден аксиален спондилоартрит), е подобна на тази, наблюдавана в проучванията при псориазис.
Неутропения
В клиничните проучвания фаза III при псориазис неутропения се наблюдава по-често при секукинумаб, отколкото при плацебо, но повечето случаи са леки, преходни и търпят обратно развитие. Неутропения <1,0-0,5x109/l (CTCAE степен 3) се съобщава при 18 от 3 430 (0,5%) пациенти на лечение със секукинумаб, при липса на зависимост от дозата и без да е свързана с поява на инфекции във времето при 15 от 18 случая. Няма съобщения за по-тежки случаи на неутропения. При останалите 3 случая се съобщава за наличие на инфекции, които не са били сериозни, повлияли са се добре от стандартната терапия и не са налагали преустановяване на лечението със секукинумаб.
Честотата на случаите на неутропения при псориатичен артрит и аксиален спондилоартрит (анкилозиращ спондилит и нерентгенографски потвърден аксиален спондилоартрит) е подобна на тази при псориазис.
Съобщават се редки случаи на неутропения <0,5x109/l (CTCAE степен 4).
Реакции на свръхчувствителност
В клиничните проучвания са наблюдавани редки случаи на уртикария и анафилактични реакции към секукинумаб (вж. също точка 4.4).
Имуногенност
В клиничните проучвания при псориазис, псориатичен артрит и аксиален спондилоартрит (анкилозиращ спондилит и нерентгенографски потвърден аксиален спондилоартрит), по-малко от 1% от пациентите на лечение със секукинумаб образуват антитела към секукинумаб в рамките на 52-седмичния период на лечение. Около половината от образуваните по време на лечението антитела срещу лекарствения продукт са били неутрализиращи, но това не е било свързано със загуба на ефикасност или отклонения във фармакокинетиката.
Педиатрична популация
Нежелани лекарствени реакции при педиатрични пациенти на възраст от 6 години нагоре с плакатен псориазис
Безопасността на секукинумаб е оценена в две проучвания фаза III при педиатрични пациенти с плакатен псориазис. Първото проучване (педиатрично проучване 1) е двойносляпо, плацебо-контролирано проучване при 162 пациенти на възраст от 6 до под 18 години с тежък плакатен псориазис. Второто проучване (педиатрично проучване 2) е открито проучване при 84 пациенти на възраст от 6 до под 18 години с умерен до тежък плакатен псориазис. Профилът на безопасност, съобщен при тези две проучвания, съответства на профила на безопасност, съобщен при възрастни пациенти с плакатен псориазис.
Нежелани лекарствени реакции при педиатрични пациенти с ЮИА
Безопасността на секукинумаб е оценена също и в проучване фаза III при 86 пациенти с ювенилен идиопатичен артрит с ERA и ЮПсА, на възраст от 2 до по-малко от 18 години. Профилът на безопасност, съобщен в това проучване, съответства на профила на безопасност, съобщен при възрастни пациенти.
Съобщаване на подозирани нежелани реакции
Съобщаването на подозирани нежелани реакции след разрешаване за употреба на лекарствения продукт е важно. Това позволява да продължи наблюдението на съотношението полза/риск за лекарствения продукт. От медицинските специалисти се изисква да съобщават всяка подозирана нежелана реакция чрез националния регулаторен орган на адрес:
България
Изпълнителна агенция по лекарствата
ул. „Дамян Груев” № 8
1303 София
Teл.: +359 2 8903417
уебсайт: www.bda.bg [2]
4.9 Предозиране
Дози до 30 mg/kg (приблизително 2 000 до 3 000 mg) са били прилагани интравенозно в клинични проучвания, без поява на дозолимитираща токсичност. В случай на предозиране се препоръчва пациентът да бъде проследен за поява на някакви признаци или симптоми на нежелани реакции и да се започне незабавно съответното симптоматично лечение.
5. ФАРМАКОЛОГИЧНИ СВОЙСТВА
5.1 Фармакодинамични свойства
Фармакотерапевтична група: Имуносупресори, инхибитори на интерлевкините, ATC код: L04AC10
Механизъм на действие
Секукинумаб е изцяло човешко IgG1/κ моноклонално антитяло, което селективно се свързва и неутрализира проинфламаторния цитокин интерлевкин-17A (IL-17A). Секукинумаб действа като се свързва с IL-17A и инхибира взаимодействието му с IL-17 рецептора, който се експресира върху различни видове клетки, включително кератиноцитите. В резултат на това секукинумаб инхибира освобождаването на проинфламаторни цитокини, хемокини и медиатори на тъканна увреда и намалява IL-17A-медиирания принос в развитието на автоимунните и възпалителните заболявания. Клинично значими нива секукинумаб достигат кожата и намаляват нивото на локалните маркери на възпаление. Като директен ефект лечението със секукинумаб намалява еритема, уплътняването и десквамацията, които са налице в лезиите при плакатен псориазис.
IL-17A е естествено образуващ се цитокин, който участва в нормалния възпалителен и имунен отговор. IL-17A играе ключова роля в патогенезата на плакатния псориазис, псориатичен артрит и аксиалния спондилоартрит (анкилозиращ спондилит и нерентгенографски потвърден аксиален спондилоартрит) и нивата му са повишени в засегнатата от лезии кожа за разлика от незасегнатата кожа при пациенти с плакатен псориазис и в синовиалната тъкан при пациентите с псориатичен артрит. Честотата на произвеждащите IL-17 клетки също така е значимо по-висока в субхондралния костен мозък на фасетните стави при пациентите с анкилозиращ спондилит. При пациентите с нерентгенографски потвърден аксиален спондилоартрит също са наблюдавани повишени нива на лимфоцити, произвеждащи IL-17A. Инхибирането на IL-17A е показало ефективност при лечението на анкилозиращ спондилит, като така се установява ключовата роля на този цитокин при аксиалния спондилоартрит.
Фармакодинамични ефекти
Серумните нива на общия IL-17A (свободен и свързан със секукинумаб IL-17A) първоначално се повишават при пациентите, получаващи секукинумаб. Следва бавно понижаване вследствие на намаления клирънс на свързания със секукинумаб IL-17A, което показва, че секукинумаб се свързва селективно със свободния IL-17A, играещ ключова роля в патогенезата на плакатния псориазис.
В проучване със секукинумаб, инфилтриращите епидермиса неутрофили и различни, свързани с неутрофилите маркери, които са повишени в кожните лезии при пациенти с плакатен псориазис, са значимо понижени след една до две седмици лечение.
Установено е, че секукинумаб понижава (в рамките на 1 до 2 седмици от лечението) нивата на С-реактивния протеин, който е маркер за наличие на възпаление.
Клинична ефикасност и безопасност
Плакатен псориазис при възрастни
Безопасността и ефикасността на секукинумаб са оценени в четири рандомизирани, двойно слепи, плацебо-контролирани проучвания фаза III при пациенти с умерен до тежък плакатен псориазис, които са кандидати за фототерапия или системна терапия [ERASURE, FIXTURE, FEATURE, JUNCTURE]. Ефикасността и безопасността на секукинумаб 150 mg и 300 mg са оценени или спрямо плацебо или спрямо етанерцепт. В допълнение, едно проучване оценява схемата на продължителна терапия спрямо схемата на „лечение при нужда“ [SCULPTURE].
От 2 403 пациенти, които са включени в плацебо-контролираните проучвания, 79% не са били лекувани до момента с биологични лекарствени продукти, при 45% е наблюдаван неуспех от терапия, която не е включвала биологични лекарствени продукти, а при 8% е наблюдаван неуспех от приложената терапия с биологични лекарствени продукти (при 6% е наблюдаван неуспех от анти-TNF, а при 2% неуспех от anti-p40). Приблизително 15 до 25% от пациентите в проучванията фаза III имат псориатичен артрит (ПсА) на изходно ниво.
Проучване при псориазис 1 (ERASURE) оценява 738 пациенти. Пациентите, рандомизирани на секукинумаб, получават дози от 150 mg или 300 mg на седмица 0, 1, 2, 3 и 4, последвани от същата доза всеки месец. Проучване при псориазис 2 (FIXTURE) оценява 1 306 пациенти. Пациентите, рандомизирани на секукинумаб, получават дози от 150 mg или 300 mg на седмица 0, 1, 2, 3 и 4, последвани от същата доза, приложена всеки месец. Пациентите, рандомизирани на етанерцепт получават доза от 50 mg два пъти седмично в продължение на 12 седмици, последвана от 50 mg всяка седмица. Както в проучване 1, така и в проучване 2, пациентите, рандомизирани на плацебо, при които не е наблюдавано повлияване на 12-та седмица преминават на секукинумаб (или 150 mg или 300 mg) на седмица 12, 13, 14 и 15, последвани от същата доза, приложена всеки месец, като се започва от седмица 16. Всички пациенти са проследени до 52 седмици след започване на лечението.
Проучване при псориазис 3 (FEATURE) оценява 177 пациенти, използващи предварително напълнената спринцовка спрямо плацебо 12 седмици след започване на лечението, като целта е да се оцени безопасността, поносимостта и ползата от самостоятелното прилагане на секукинумаб чрез предварително напълнена спринцовка. Проучване при псориазис 4 (JUNCTURE) оценява 182 пациенти, използващи предварително напълнена писалка спрямо плацебо 12 седмици след започване на лечението, като целта е да се оцени безопасността, поносимостта и ползата от самостоятелното прилагане на секукинумаб чрез предварително напълнена писалка. Както в проучване 3, така и в проучване 4, пациентите, рандомизирани на секукинумаб, получават дози от 150 mg или 300 mg на седмица 0, 1, 2, 3 и 4, последвани от същата доза, приложена всеки месец. Пациентите са рандомизирани също така да получават плацебо на седмица 0, 1, 2, 3 и 4, последвано от същата доза, приложена всеки месец.
Проучване при псориазис 5 (SCULPTURE) оценява 966 пациенти. Всички пациенти получават секукинумаб в дози от 150 mg или 300 mg на седмица 0, 1, 2, 3, 4, 8 и 12, след което са рандомизирани на поддържаща схема, да получават същата доза всеки месец, започвайки от седмица 12 или на схема „лечение при нужда“ в същата доза. Пациентите, рандомизирани на схема „лечение при нужда“ не постигат задоволително поддържане на терапевтичния отговор, поради което се препоръчва фиксираната ежемесечна поддържаща схема.
Първичните съставни крайни точки в плацебо и активно контролираните проучвания са процентът пациенти, постигнали терапевтичен отговор PASI 75 и IGA mod 2011 „чиста“ или „почти чиста“ кожа спрямо плацебо на 12-та седмица (вж. Таблици 4 и 5). Дозата от 300 mg подобрява състоянието на кожата до „чиста“ или „почти чиста“ в крайните точки за ефикасност PASI 90, PASI 100, и IGA mod 2011 0 или 1 във всички проучвания с най-голям ефект на седмица 16, поради тази причина се препоръчва тази доза.
Таблица 4 Обобщение на клиничен отговор PASI 50/75/90/100 & IGA⃰ mod 2011 „чиста“ или „почти чиста“ кожа в проучвания при псориазис 1, 3 и 4 (ERASURE, FEATURE и JUNCTURE)
|
Седмица 12 |
|
Седмица 16 |
Седмица 52 |
||||
|
|
Плацебо |
150 mg |
300 mg |
150 mg |
300 mg |
150 mg |
300 mg |
|
Проучване 1 |
|
|
|
|
|
|
|
|
Брой пациенти |
246 |
244 |
245 |
244 |
245 |
244 |
245 |
|
PASI 50 отговор n (%) |
22 (8,9%) |
203 (83,5%) |
222 (90,6%) |
212 (87,2%) |
224 (91,4%) |
187 (77%) |
207 (84,5%) |
|
PASI 75 отговор n (%) |
11 (4,5%) |
174 (71,6%)** |
200 (81,6%)** |
188 (77,4%) |
211 (86,1%) |
146 (60,1%) |
182 (74,3%) |
|
PASI 90 отговор n (%) |
3 (1,2%) |
95 (39,1%)** |
145 (59,2%)** |
130 (53,5%) |
171 (69,8%) |
88 (36,2%) |
147 (60,0%) |
|
PASI 100 отговор n (%) |
2 (0,8%) |
31 (12,8%) |
70 (28,6%) |
51 (21,0%) |
102 (41,6%) |
49 (20,2%) |
96 (39,2%) |
|
IGA mod 2011 „чиста“ или „почти чиста“ n (%) |
6 (2,40%) |
125 (51,2%)** |
160 (65,3%)** |
142 (58,2%) |
180 (73,5%) |
101 (41,4%) |
148 (60,4%) |
|
Проучване 3 |
|
|
|
|
|
|
|
|
Брой пациенти |
59 |
59 |
58 |
- |
- |
- |
- |
|
PASI 50 отговор n (%) |
3 (5,1%) |
51 (86,4%) |
51 (87,9%) |
- |
- |
- |
- |
|
PASI 75 отговор n (%) |
0 (0,0%) |
41 (69,5%)** |
44 (75,9%)** |
- |
- |
- |
- |
|
PASI 90 отговор n (%) |
0 (0,0%) |
27 (45,8%) |
35 (60,3%) |
- |
- |
- |
- |
|
PASI 100 отговор n (%) |
0 (0,0%) |
5 (8,5%) |
25 (43,1%) |
- |
- |
- |
- |
|
IGA mod 2011 „чиста“ или „почти чиста“ n (%) |
0 (0,0%) |
31 (52,5%)** |
40 (69,0%)** |
- |
- |
- |
- |
|
Проучване 4 |
|
|
|
|
|
|
|
|
Брой пациенти |
61 |
60 |
60 |
- |
- |
- |
- |
|
PASI 50 отговор n (%) |
5 (8,2%) |
48 (80,0%) |
58 (96,7%) |
- |
- |
- |
- |
|
PASI 75 отговор n (%) |
2 (3,3%) |
43 (71,7%)** |
52 (86,7%)** |
- |
- |
- |
- |
|
PASI 90 отговор n (%) |
0 (0,0%) |
24 (40,0%) |
33 (55,0%) |
- |
- |
- |
- |
|
PASI 100 отговор n(%) |
0 (0,0%) |
10 (16,7%) |
16 (26,7%) |
- |
- |
- |
- |
|
IGA mod 2011 „чиста“ или „почти чиста“ n (%) |
0 (0,0%) |
32 (53,3%)** |
44 (73,3%)** |
- |
- |
- |
- |
|
* IGA mod 2011 е 5-степенна категорийна скала, включваща „0 = чиста“, „1 = почти чиста“, „2 = лек“, „3 = умерен“ или „4 = тежък“, показваща цялостната оценка на лекаря за тежестта на псориазиса, фокусирайки се върху индурацията, еритема и беленето на кожата. Ефект от лечението, дефиниран като „чиста“ или „почти чиста“ кожа представлява липса на признаци на псориазис или нормално до розово оцветяване на лезиите, липса на задебеляване на плаките и липса или минимално локално белене на кожата. ** p стойности спрямо плацебо и коригирани за множественост: p<0,0001. |
|||||||
Таблица 5 Обобщение на клиничния отговор в проучване при псориазис 2 (FIXTURE)
|
Седмица 12 |
|
|
Седмица 16 |
|
|
Седмица 52 |
||||
|
|
Плацебо |
150 mg |
300 mg |
Етанерце пт |
150 mg |
300 mg |
Етанерцепт |
150 mg |
300 mg |
Етанерцепт |
|
Брой пациенти |
324 |
327 |
323 |
323 |
327 |
323 |
323 |
327 |
323 |
323 |
|
PASI 50 отговор n (%) |
49 (15,1%) |
266 (81,3%) |
296 (91,6%) |
226 (70,0%) |
290 (88,7%) |
302 (93,5%) |
257 (79,6%) |
249 (76,1%) |
274 (84,8%) |
234 (72,4%) |
|
PASI 75 отговор n (%) |
16 (4,9%) |
219 (67,0%) ** |
249 (77,1%) ** |
142 (44,0%) |
247 (75,5%) |
280 (86,7%) |
189 (58,5%) |
215 (65,7%) |
254 (78,6%) |
179 (55,4%) |
|
PASI 90 отговор n (%) |
5 (1,5%) |
137 (41,9%) |
175 (54,2%) |
67 (20,7%) |
176 (53,8%) |
234 (72,4%) |
101 (31,3%) |
147 (45,0%) |
210 (65,0%) |
108 (33,4%) |
|
PASI 100 отговор n (%) |
0 (0%) |
47 (14,4%) |
78 (24,1%) |
14 (4,3%) |
84 (25,7%) |
119 (36,8%) |
24 (7,4%) |
65 (19,9%) |
117 (36,2%) |
32 (9,9%) |
|
IGA mod 2011 „чиста“ или „почти чиста“ n (%) |
9 (2,8%) |
167 (51,1%) ** |
202 (62,5%) ** |
88 (27,2%) |
200 (61,2%) |
244 (75,5%) |
127 (39,3%) |
168 (51,4%) |
219 (67,8%) |
120 (37,2%) |
В допълнително проучване при псориазис (CLEAR) са оценени 676 пациенти. Секукинумаб 300 mg постига първичната и вторичната крайна точка, показвайки превъзходство спрямо устекинумаб по отношение на PASI 90 отговора на седмица 16 (първична крайна точка), бързата поява на PASI 75 отговор на седмица 4 и дългосрочен PASI 90 отговор на седмица 52. По-голямата ефикасност на секукинумаб спрямо устекинумаб по отношение на крайните точки PASI 75/90/100 и IGA mod 2011 0 или 1 („чиста“ или „почти чиста“) се наблюдава рано и продължава през цялото време до седмица 52.
Таблица 6 Обобщение на клиничния отговор в проучването CLEAR
|
Седмица 4 |
Седмица 16 |
Седмица 52 |
||||
|
|
Секукинумаб 300 mg |
Устекинумаб* |
Секукинумаб 300 mg |
Устекинумаб* |
Секукинумаб 300 mg |
Устекинумаб* |
|
Брой пациенти |
334 |
335 |
334 |
335 |
334 |
335 |
|
PASI 75 отговор n (%) |
166 (49,7%)** |
69 (20,6%) |
311 (93,1%) |
276 (82,4%) |
306 (91,6%) |
262 (78,2%) |
|
PASI 90 отговор n (%) |
70 (21,0%) |
18 (5,4%) |
264 (79,0%)** |
192 (57,3%) |
250 (74,9%)*** |
203 (60,6%) |
|
PASI 100 отговор n (%) |
14 (4,2%) |
3 (0,9%) |
148 (44,3%) |
95 (28,4%) |
150 (44,9%) |
123 (36,7%) |
|
IGA mod 2011 „чиста“ или „почти чиста“ n (%) |
128 (38,3%) |
41 (12,2%) |
278 (83,2%) |
226 (67,5%) |
261 (78,1%) |
213 (63,6%) |
* Пациентите, лекувани със секукинумаб, получават 300 mg на седмица 0, 1, 2, 3 и 4, последвани от същата доза на всеки 4 седмици до седмица 52. Пациентите, лекувани с устекинумаб, получават 45 mg или 90 mg на седмица 0 и 4, след което на всеки 12 седмици до седмица 52 (в зависимост от телесното тегло, съгласно одобрения начина на приложение)
** p стойности спрямо устекинумаб: p<0,0001 за първичната крайна точка PASI 90 на седмица 16 и вторичната крайна точка PASI 75 на седмица 4.
*** p стойности спрямо устекинумаб: p=0,0001 за вторичната крайна точка PASI 90 на седмица 52
Секукинумаб е ефикасен при пациенти, нелекувани до момента със системна терапия и биологични лекарствени продукти, пациенти, с експозиция на биологични лекарствени продукти/анти-TNF терапия и пациенти, неповлияли се от биологични лекарствени продукти/анти-TNF терапия. Подобрението в PASI 75 при пациентите със съпътстващ псориатичен артрит на изходно ниво е подобно на това в общата популация с плакатен псориазис.
Секукинумаб е свързан с бърза поява на ефикасност с 50% понижение в средния PASI на седмица 3 при доза от 300 mg.
Фигура 1 Промяна в течение на времето в процента на средния PASI скор спрямо изходната стойност в проучване 1 (ERASURE)
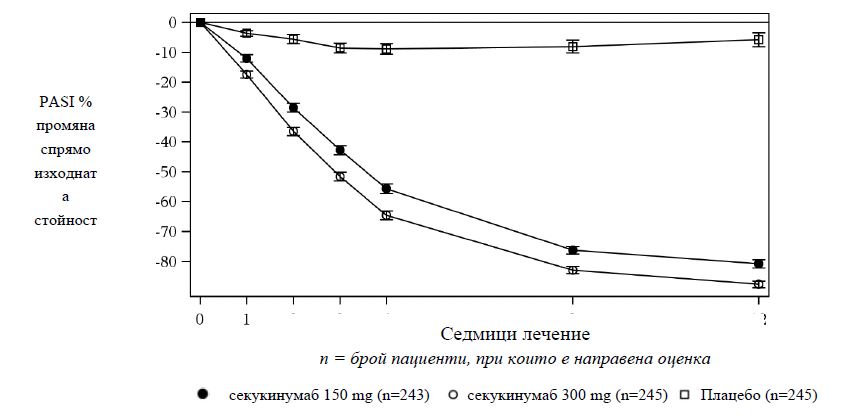
Специфични локализации/ форми на плакатен псориазис
В две допълнителни плацебо контролирани проучвания се наблюдава подобрение както при псориазиса по ноктите (TRANSFIGURE, 198 пациенти), така и при палмоплантарния плакатен псориазис (GESTURE, 205 пациенти). В проучването TRANSFIGURE секукинумаб превъзхожда плацебо на седмица 16 (46,1% за 300 mg, 38,4% за 150 mg и 11,7% за плацебо) въз основа на установеното значително подобрение спрямо изходното ниво в Индекса, оценяващ тежестта на псориазиса по ноктите - Nail Psoriasis Severity Index (NAPSI %) при пациенти с умерен до тежък плакатен псориазис със засягане на ноктите. В проучването GESTURE секукинумаб превъзхожда плацебо на седмица 16 (33,3% за 300 mg, 22,1% за 150 mg и 1,5% за плацебо) въз основа на установеното значително подобрение на ррIGA 0 или 1 („чиста“ или „почти чиста“) при пациенти с умерен до тежък палмоплантарен псориазис.
Плацебо контролирано проучване оценява 102 пациенти с умерено тежък до тежък псориазис на скалпа, дефинирани със скор по като Индекс за тежест на псориазиса по скалпа (Psoriasis Scalp Severity Index - PSSI) ≥12, IGA mod 2011 скор само за скалпа 3 или повече и засягане на поне 30% от повърхността на скалпа. Секукинумаб 300 mg превъзхожда плацебо на седмица 12 въз основа на установеното значимо подобрение при отговора, оценен със скор по PSSI 90 (52,9% спрямо 2,0%), така и при отговора IGA mod 2011 0 или 1 само за скалпа (56,9% спрямо 5,9%). Подобрението по отношение на двете крайни точки се запазва при пациентите на секукинумаб, които продължават лечението до седмица 24.
Качество на живот/съобщени от пациентите резултати
Налице е статистически значимо подобрение на седмица 12 (проучвания 1-4) спрямо изходната стойност сравнено с плацебо в DLQI (Dermatology Life Quality Index-Дерматологичен индекс за качество на живот). Средното понижение (подобрение) в DLQI спрямо изходната стойност варира от -10,4 до -11,6 при секукинумаб 300 mg, от -7,7 до -10,1 при секукинумаб 150 mg, спрямо -1,1 до -1,9 при плацебо на седмица 12. Това подобрение се запазва в продължение на 52 седмици (проучвания 1 и 2).
Четиридесет процента от участниците в проучвания 1 и 2 са попълвали Дневник на симптомите при псориазис (Psoriasis Symptom Diary©). При участниците, попълвали Дневника във всяко едно от тези проучвания, на седмица 12 се наблюдава статистически значимо подобрение спрямо изходната стойност в съобщаваните от пациентите признаци и симптомите на сърбеж,болка и белене на кожата.
Установява се статистически значимо подобрение в DLQI на седмица 4 спрямо изходното ниво при пациентите на лечение със секукинумаб спрямо пациентите на лечение с устекинумаб (CLEAR) и това подобрение се запазва до седмица 52.
Установява се статистически значимо подобрение на съобщаваните от пациентите признаци и симптоми на сърбеж, болка и белене на кожата, попълнени в Psoriasis Symptom Diary©, на седмица 16 и седмица 52 (CLEAR) при пациентите на лечение със секукинумаб спрямо пациентите на лечение с устекинумаб.
В проучването при псориазис на скалпа, на седмица 12 спрямо изходното ниво, се установява статистически значимо подобрение (намаляване) на съобщаваните от пациентите признаци и симптоми на сърбеж на скалпа, болка и белене на кожата в сравнение с плацебо.
Гъвкавост на дозиране при плакатен псориазис
Рандомизирано, двойносляпо, многоцентрово проучване оценява две поддържащи схеми на прилагане (300 mg на всеки 2 седмици [Q2W] и 300 mg на всеки 4 седмици [Q4W]), прилагани като 150 mg предварително напълнена спринцовка при 331 пациенти с телесно тегло ≥90 kg, с умерен до тежък псориазис. Пациентите са рандомизирани 1:1, както следва:
• секукинумаб 300 mg на седмица 0, 1, 2, 3 и 4, последвано от същата доза на всеки 2 седмици (Q2W) до седмица 52 (n=165);
• секукинумаб 300 mg на седмица 0, 1, 2, 3 и 4, последвано от същата доза на всеки 4 седмици (Q4W) до седмица 16 (n=166);
o Пациентите, рандомизирани да получават секукинумаб 300 mg Q4W, които са постигнали отговор PASI 90 на седмица 16, са продължили да получават същата схема на прилагане до седмица 52. Пациентите, рандомизирани да получават секукинумаб 300 mg Q4W, които не са постигнали отговор PASI 90 на седмица 16, са продължили или със същата схема на прилагане, или са пренасочени да получават секукинумаб 300 mg Q2W до седмица 52.
Общо, честотата на отговора за ефикасност в групата, лекувана по схема на всеки 2 седмици, е по-висока, в сравнение с групата, лекувана по схема на всеки 4 седмици (Таблица 7).
Таблица 7 Обобщение на клиничния отговор в проучването за гъвкавост на дозирането при плакатен псориазис*
|
|
Седмица 16 |
Седмица 52 |
||
|
секукинумаб 300 mg Q2W |
секукинумаб 300 mg Q4W |
секукинумаб 300 mg Q2W |
секукинумаб 300 mg Q4W1 |
|
|
Брой пациенти |
165 |
166 |
165 |
83 |
|
Отговор PASI 90, n (%) |
121 (73,2%) ** |
92 (55,5%) |
126 (76,4%) |
44 (52,4%) |
|
Отговор IGA mod 2011 „чиста” или „почти чиста” кожа, n (%) |
122 (74,2%)2 |
109 (65,9%)2 |
125 (75,9%) |
46 (55,6%) |
|
* Множествено приписване на стойности 1 300 mg Q4W:пациенти, лекувани продължително с 300 mg Q4W, независимо от статуса на отговора PASI 90 на седмица 16; 43 пациенти са постигнали отговор PASI 90 на седмица 16, а 40 пациенти не са постигнали отговор PASI 90 на седмица 16. ** Едностранна p стойност = 0,0003 за първична крайна точка PASI 90 на седмица 16. 2 Няма статистическа значимост. |
||||
При пациентите, които не са постигнали отговор PASI 90 на седмица 16, и са титрирани възходящо до секукинумаб
300 mg Q2W, честотата на отговор PASI 90 се подобрява, в сравнение с тези, които са останали на схема на прилагане на секукинумаб 300 mg Q4W, докато честотата на отговор IGA mod 2011 0/1 остава постоянна във времето и в двете групи на лечение.
Профилите на безопасност на двете схеми на прилагане, Cosentyx 300 mg прилаган на всеки 4 седмици и Cosentyx 300 mg прилаган на всеки 2 седмици, при пациенти с телесно тегло ≥90 kg, са сравними и съответстват на профила на безопасност, съобщаван при пациенти с псориазис.
Псориатичен артрит
Безопасността и ефикасността на секукинумаб са оценени при 1 999 пациенти в три рандомизирани, двойнослепи, плацебо-контролирани проучвания фаза III при пациенти с активен псориатичен артрит (≥3 оточни и ≥3 болезнени стави), въпреки провежданото лечение с нестероидни противовъзпалителни средства (НСПВС), кортикостероиди или модифициращи болестта антиревматоидни средства (DMARD). В проучванията са включени пациенти с ПсА от всеки подвид, включително полиартикуларен артрит без данни за ревматоидни възли, спондилит с периферен артрит, асиметричен периферен артрит, ангажиране на дисталните интерфалангеални стави и мутилиращ артрит. Пациентите в проучванията са с диагностициран ПсА от поне пет години. Мнозинството от пациентите също така имат активни псориатични кожни лезии или документирана анамнеза за наличие на псориазис. Над 61% и 42% от пациентите с ПсА са имали съответно ентезит и дактилит на изходно ниво. Във всички проучвания първична крайна точка е постигнат отговор 20 по критериите на Американския колеж по ревматология (American College of Rheumatology - ACR). В проучването при псориатичен артрит 1 (проучване при ПсА 1) и проучването при псориатичен артрит 2 (проучване при ПсА 2) първичната крайна точка се оценява на седмица 24. В проучването при псориатичен артрит 3 (проучване при ПсА 3) първичната крайна точка, заедно с основните вторични крайни точки се оценяват на седмица 16, а промяната спрямо изходното ниво в модифицирания общ скор по Sharp (modified Total Sharp Score, mTSS) – на седмица 24.
В проучване при ПсА 1, проучване при ПсА 2 и проучване при ПсА 3 съответно 29%, 35% и 30% от пациентите са лекувани преди това с анти-TNFα средство и са преустановили лечението с анти-TNFα средството поради липса на ефект или непоносимост (пациенти с незадоволителен отговор към анти-TNFα).
Проучване при ПсА 1 (FUTURE 1) оценява 606 пациенти, от които 60,7% провеждат съпътстващо лечение с MTX. При пациентите, рандомизирани на секукинумаб, се прилагат 10 mg/kg интравенозно на седмица 0, 2 и 4, последвани от 75 mg или 150 mg подкожно всеки месец, като се започне от седмица 8. Пациентите, рандомизирани на плацебо, при които не се наблюдава терапевтичен отговор на седмица 16 (ранна спасителна терапия), и другите пациенти на лечение с плацебо, на седмица 24 преминават на лечение със секукинумаб (75 mg или 150 mg подкожно), последвани от същата доза всеки месец.
Проучване при ПсА 2 (FUTURE 2) оценява 397 пациенти, от които 46,6% провеждат съпътстващо лечение с MTX. При пациентите, рандомизирани на секукинумаб, се прилагат 75 mg, 150 mg или 300 mg подкожно на седмица 0, 1, 2, 3 и 4, последвани от същата доза всеки месец. Пациентите, рандомизирани на плацебо, при които не се наблюдава терапевтичен отговор на седмица 16 (ранна спасителна терапия), преминават на лечение със секукинумаб (150 mg или 300 mg подкожно) на седмица 16, последвани от същата доза всеки месец. Пациентите, рандомизирани на плацебо, при които се наблюдава терапевтичен отговор на седмица 16, преминават на лечение със секукинумаб (150 mg или 300 mg подкожно) на седмица 24, последвани от същата доза всеки месец.
Проучване при ПсА 3 (FUTURE 5) оценява 996 пациенти, от които 50,1% провеждат съпътстващо лечение с MTX. Пациентите са рандомизирани да получават секукинумаб 150 mg, 300 mg или плацебо, приложени подкожно на седмица 0, 1, 2, 3 и 4, последвани от същата доза всеки месец, или инжекция секукинумаб 150 mg веднъж месечно (без натоварване).
Пациентите, рандомизирани да получават плацебо, при които не се наблюдава терапевтичен отговор на седмица 16 (ранна спасителна терапия) преминават на лечение със секукинумаб (150 mg или 300 mg подкожно) на седмица 16, последвани от същата доза всеки месец. Пациентите, рандомизирани да получават плацебо, при които се наблюдава терапевтичен отговор на седмица 16, преминават на лечение със секукинумаб (150 mg или 300 mg подкожно) на седмица 24, последвани от същата доза всеки месец.
Признаци и симптоми
Лечението със секукинумаб води до значимо подобрение на показателите за активност на заболяването спрямо плацебо на седмици 16 и 24 (вж. Таблица 8).
Таблица 8 Клиничен отговор в проучване при ПсА 2 и проучване при ПсА 3 на седмица 16 и седмица 24
|
|
Проучване при ПсА 2 |
Проучване при ПсА 3 |
||||
|
|
Плацебо |
150 mg1 |
300 mg1 |
Плацебо |
150 mg1 |
300 mg1 |
|
Брой рандомизирани пациенти |
98 |
100 |
100 |
332 |
220 |
222 |
|
ACR20 отговор n (%) |
|
|
|
|
|
|
|
Седмица 16 |
18 (18,4%) |
60 (60,0%***) |
57 (57,0%***) |
91◊ (27,4%) |
122◊ (55,5%***) |
139◊ (62,6%***) |
|
Седмица 24 |
15◊ (15,3%) |
51◊ (51,0%***) |
54◊ (54,0%***) |
78 (23,5%) |
117 (53,2%***) |
141 (63,5%***) |
|
ACR50 отговор n (%) |
|
|
|
|
|
|
|
Седмица 16 |
6 (6,1%) |
37 (37,0%***) |
35 (35,0%***) |
27 (8,1%) |
79 (35,9%*) |
88 (39,6%*) |
|
Седмица 24 |
7 (7,1%) |
35 (35,0%) |
35 (35,0%**) |
29 (8,7%) |
86 (39,1%***) |
97 (43,7%***) |
|
ACR70 отговор n (%) |
|
|
|
|
|
|
|
Седмица 16 |
2 (2,0%) |
17 (17,0%**) |
15 (15,0%**) |
14 (4,2%) |
40 (18,2%***) |
45 (20,3%***) |
|
Седмица 24 |
1 (1,0%) |
21 (21,0%**) |
20 (20,0%**) |
13 (3,9%) |
53 (24,1%***) |
57 (25,7%***) |
|
DAS28-CRP |
|
|
|
|
|
|
|
Седмица 16 |
-0,50 |
-1,45*** |
-1,51*** |
-0,63 |
-1,29* |
-1,49* |
|
Седмица 24 |
-0,96 |
-1,58** |
-1,61** |
-0,84 |
-1,57*** |
-1,68*** |
|
Брой пациенти с ангажиране на ≥3% от телесната повърхност (BSA) с псориатични лезии на изходно ниво |
43 (43,9%) |
58 (58,0%) |
41 (41,0%) |
162 (48,8%) |
125 (56,8%) |
110 (49,5%) |
|
PASI 75 отговор n (%) |
|
|
|
|
|
|
|
Седмица 16 |
3 (7,0%) |
33 (56,9%***) |
27 (65,9%***) |
20 (12,3%) |
75 (60,0%*) |
77 (70,0%*) |
|
Седмица 24 |
7 (16,3%) |
28 (48,3%**) |
26 (63,4%***) |
29 (17,9%) |
80 (64,0%***) |
78 (70,9%***) |
|
PASI 90 отговор n (%) |
|
|
|
|
|
|
|
Седмица 16 |
3 (7,0%) |
22 (37,9%***) |
18 (43,9%***) |
15 (9,3%) |
46 (36,8%*) |
59 (53,6%*) |
|
Седмица 24 |
4 (9,3%) |
19 (32,8%**) |
20 (48,8%***) |
19 (11,7%) |
51 (40,8%***) |
60 (54,5%***) |
|
Обратно развитие на дактилита n (%) † |
|
|
|
|
|
|
|
Седмица 16 |
10 (37%) |
21 (65,6%*) |
26 (56,5%) |
40 (32,3%) |
46 (57,5%*) |
54 (65,9%*) |
|
Седмица 24 |
4 (14,8%) |
16 (50,0%**) |
26 (56,5%**) |
42 (33,9%) |
51 (63,8%***) |
52 (63,4%***) |
|
Обратно развитие на ентезита n (%) ‡ |
|
|
|
|
|
|
|
Седмица 16 |
17 (26,2%) |
32 (50,0%**) |
32 (57,1%***) |
68 (35,4%) |
77 (54,6%*) |
78 (55,7%*) |
|
Седмица 24 |
14 (21,5%) |
27 (42,2%*) |
27 (48,2%**) |
66 (34,4%) |
77 (54,6%***) |
86 (61,4%***) |
|
* p<0,05, ** p<0,01, *** p<0,001; спрямо плацебо Всички p-стойности са коригирани за множественост на изследването въз основа на предварително определена йерархия на седмица 24 в проучване при ПсА 2, с изключение на тези за ACR70, дактилит и ентезит, които са експлораторни крайни точки и всички крайни точки на седмица 16. Всички p-стойности са коригирани за множественост на изследването въз основа на предварително определена йерархия на седмица 16 в проучване при ПсА 3, с изключение на тези за ACR70, която е експлораторна крайна точка и всички крайни точки на седмица 24. Приписана стойност при липса на отговор при липсваща бинарна крайна точка. ACR: Американския колеж по ревматология; PASI: индекс на засегнат участък и тежест на псориазиса; DAS: скор за активност на заболяването; BSA: телесна повърхност
1Секукинумаб 150 mg или 300 mg s.c. на седмица 0, 1, 2, 3 и 4, последвани от същата доза всеки месец †При пациенти с дактилит на изходно ниво (n=27, 32, 46, съответно в проучване при ПсА 2 и n=124, 80, 82, съответно в изпитване при ПсА 3) ‡ При пациенти с ентезит на изходно ниво (n=65, 64, 56, съответно в проучване при ПсА 2 и n=192, 141, 140, съответно в изпитване при ПсА 3) |
||||||
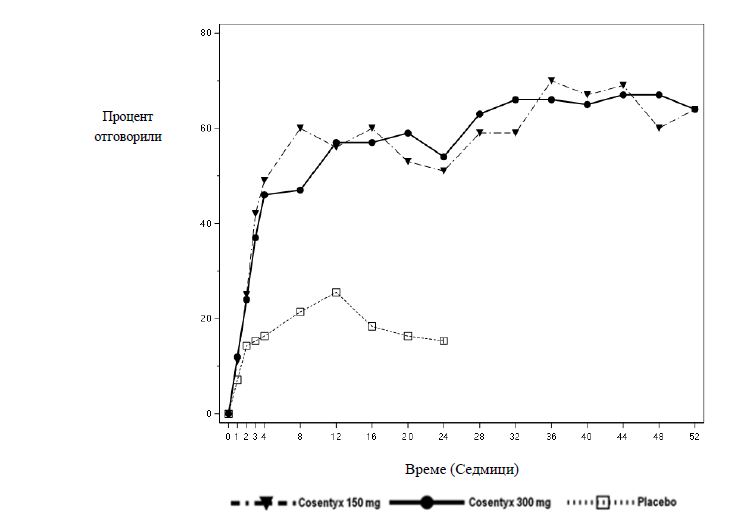
Секукинумаб започва да действа на седмица 2. Статистически значима разлика в ACR 20 спрямо плацебо се постига на седмица 3.
Процентът на пациентите, постигнали клиничен отговор според критериите на ACR 20 при съответните визити, е показан на Фигура 2.
Фигура 2 Клиничен отговор според критериите на ACR20 в проучване при ПсА 2 в течение на времето до седмица 52
При пациентите с ПсА се наблюдава подобен отговор по отношение на първичната и основните вторични крайни точки, независимо от това, дали са на съпътстващо лечение с MTX или не. В проучване при ПсА 2 на седмица 24 пациентите, лекувани със секукинумаб, провеждащи съпътстващо лечение с MTX, имат по-висок процент на постигнат терапевтичен отговор според критериите на ACR 20 (47,7% и 54,4% съответно при 150 mg и 300 mg, спрямо плацебо 20,0%) и ACR 50 (31,8% и 38,6% съответно при 150 mg и 300 mg, спрямо плацебо 8,0%). Пациентите, лекувани със секукинумаб, без съпътстващо лечение с MTX имат по-висок процент на постигнат терапевтичен отговор според критериите на ACR 20 (53,6% и 53,6% съответно при 150 mg и 300 mg спрямо плацебо 10,4%) и ACR 50 (37,5% и 32,1% съответно при 150 mg и 300 mg спрямо плацебо 6,3%).
В проучване при ПсА 2 както пациентите, при които не е провеждана анти-TNFα терапия, така и пациентите с незадоволителен отговор към анти-TNFα терапия, лекувани със секукинумаб, имат значително по-висок процент на постигнат терапевтичен отговор според критериите на ACR 20 спрямо плацебо на седмица 24, като в групата, при която не е провеждана анти-TNFα терапия се наблюдава малко по-висок процент на постигнат терапевтичен отговор (пациенти, при които не е провеждана анти-TNFα терапия: 64% и 58% съответно при 150 mg и 300 mg спрямо плацебо 15,9%; пациенти с незадоволителен отговор към анти-TNFα: 30% и 46% съответно при 150 mg и 300 mg спрямо плацебо 14,3%). В подгрупата пациенти с незадоволителен отговор към анти-TNFα, само дозата от 300 mg показва значимо по-висок процент на постигнат терапевтичен отговор според критериите на ACR 20 спрямо плацебо (p<0,05) и показва клинично значимо превъзходство спрямо 150 mg по отношение на множеството вторични крайни точки. Подобрение в PASI 75 отговора се наблюдава и в двете подгрупи, като дозата от 300 mg показва статистически значим благоприятен ефект при пациентите с незадоволителен отговор към анти-TNFα.
Подобрение се наблюдава във всички компоненти на ACR скоровете, включително оценката на болката от страна на пациентите. В проучване при ПсА 2 процентът пациенти, постигнали терапевтичен отговор според модифицираните критерии за терапевтичен отговор при ПсА, е по-висок при пациентите, лекувани със секукинумаб (59,0% и 61,0% съответно при 150 mg и 300 mg) спрямо плацебо (26,5%) на седмица 24.
В проучване при ПсА 1 и проучване при ПсА 2 ефикасността се запазва до седмица 104. В проучване при ПсА 2 от 200 пациенти, рандомизирани първоначално да получават секукинумаб 150 mg и 300 mg, 178 (89%) пациенти продължават да бъдат на това лечение на седмица 52. От 100 пациенти, рандомизирани на секукинумаб 150 mg, 64, 39 и 20 имат съответно ACR 20/50/70 терапевтичен отговор. От 100 пациенти, рандомизирани на секукинумаб 300 mg, 64, 44 и 24 имат съответно ACR 20/50/70 терапевтичен отговор.
Рентгенологичен отговор
В проучване при ПсА 3, инхибирането на прогресията на структурното увреждане е оценено рентгенологично и е представено като промяна в модифицирания общ скор по Sharp (modified Total Sharp Score, mTSS) и неговите компоненти, скора за ерозиите (Erosion Score, ES) и скора за стесняване на ставната цепка (Joint Space Narrowing score, JSN). Направени са рентгенографии на длани, китки и стъпала на изходно ниво, седмица 16 и/или седмица 24, които са оценени независимо от поне двама човека, които са били заслепени по отношение на терапевтичната група и поредността на визитата. Лечението със секукинумаб 150 mg и 300 mg значимо инхибира скоростта на прогресия на периферното ставно увреждане в сравнение с плацебо, според измерената промяна спрямо изходното ниво в mTSS на седмица 24 (Таблица 9).
Инхибирането на прогресията на структурното увреждане е оценено също така в проучване при ПсА 1 на седмица 24 и 52, спрямо изходното ниво. Данните от седмица 24 са представени в Таблица 9.
Таблица 9 Промяна в модифицирания общ скор по Sharp при псориатичен артрит
|
|
|
Проучване при ПсА 3 |
Проучване при ПсА 1 |
||
|
Плацебо n=296 |
секукинумаб 150 mg1 n=213 |
секукинумаб 300 mg1 n=217 |
Плацебо n=179 |
секукинумаб 150 mg2 n=185 |
|
|
Общ скор |
|||||
|
Изходно ниво (SD) |
15,0 (38,2) |
13,5 (25,6) |
12,9 (23,8) |
28,4 (63,5) |
22,3 (48,0) |
|
Средна промяна на седмица 24 |
0,50 |
0,13* |
0,02* |
0,57 |
0,13* |
|
*p<0,05 въз основа на номинална, но некоригирана p-стойност 1секукинумаб 150 mg или 300 mg s.c. на седмица 0, 1, 2, 3 и 4, последвани от същата доза всеки месец 210 mg/kg на седмица 0, 2 и 4, последвани от подкожно прилагане на дози от 75 mg или 150 mg |
|||||
В проучване при ПсА 1 инхибирането на структурното увреждане се запазва при лечение със секукинумаб до седмица 52.
В проучване при ПсА 3 процентът на пациентите, при които не се наблюдава прогресия на заболяването (дефинирана като промяна спрямо изходната стойност в mTSS от ≤0,5) от рандомизацията до седмица 24 е 80,3%, 88,5% и 73,6% съответно за секукинумаб 150 mg, 300 mg и плацебо. Влияние върху инхибирането на структурното увреждане се наблюдава при пациентите, при които не е провеждана анти-TNFα терапия и пациенти с незадоволителен отговор към анти-TNFα и при пациентите със и без съпътстващо лечение с MTX.
В проучване при ПсА 1 процентът на пациентите, при които не се наблюдава прогресия на заболяването (дефинирана като промяна спрямо изходната стойност в mTSS от ≤0,5) от рандомизацията до седмица 24, е 82,3% в групата на секукинумаб, приложен като натоварваща доза от 10 mg/kg интравенозно, а след това като поддържаща доза от 150 mg подкожно и 75,7% в групата на плацебо. Процентът на пациентите, при които не се наблюдава прогресия на заболяването от седмица 24 до седмица 52, при прилагане на секукинумаб, като натоварваща доза от 10 mg/kg интравенозно, а след това като поддържаща доза от 150 mg подкожно или при пациентите на плацебо, преминали към 75 mg или 150 mg подкожно на всеки 4 седмици, на седмица 16 или седмица 24, е съответно 85,7% и 86,8%.
Аксиални прояви при ПсА
В рандомизирано, двойносляпо, плацебо-контролирано проучване (MAXIMISE) е оценена ефикасността на секукинумаб при 485 пациенти с ПсА с аксиални прояви, които не са лекувани до преди това с биологични лекарствени продукти и са имали недостатъчен отговор към лечение с НСПВС. Първичната променлива от поне 20% подобрение според критериите на Международното дружество за оценка на спондилоартрита (Assessment of SpondyloArthritis International Society - ASAS 20) на седмица 12 е изпълнена. Лечението със секукинумаб 300 mg и 150 mg в сравнение с плацебо също така води до по-голямо подобрение в признаците и симптомите (включително намаляване на болката в гръбначния стълб спрямо изходно ниво) и подобрение на физическата активност (вж. Таблица 10).
Таблица 10 Клиничен отговор в проучване MAXIMISE на седмица 12
|
|
Плацебо (n=164) |
150 mg (n=157) |
300 mg (n=164) |
|
ASAS 20 отговор, % (95% CI) |
31,2 (24,6; 38,7) |
66,3 (58,4; 73,3)* |
62,9 (55,2; 70,0)* |
|
ASAS 40 отговор, % (95% CI) |
12,2 (7,8; 18,4) |
39,5 (32,1; 47,4)** |
43,6 (36,2; 51,3)** |
|
BASDAI 50, % (95% CI) |
9,8 (5,9; 15,6) |
32,7 (25,8; 40,5)** |
37,4 (30,1; 45,4)** |
|
Болка в гръбначния стълб, VAS (95% CI) |
-13,6 (-17,2; -10,0) |
-28,5 (-32,2; -24,8)** |
-26,5 (-30,1; -22,9)** |
|
Физическа активност, HAQ-DI (95% CI) |
-0,155 (-0,224; -0,086) |
-0,330 (-0,401; -0,259)** |
-0,389 (-0,458; -0,320)** |
|
* p<0.0001; спрямо плацебо чрез множество приписани стойности. ** Сравнението спрямо плацебо не е коригирано за множественост. ASAS: Критерии на Международното дружество за оценка на спондилоартрита; BASDAI: Индекс за активност на заболяването анкилозиращ спондилит по Бат; VAS: Визуална аналогова скала; HAQ-DI (Health Assessment Questionnaire – Disability Index): Въпросник за оценка на здравето с показател за инвалидизиране. |
|||
Подобрение в ASAS 20 и ASAS 40 и за двете дози секукинумаб се наблюдава на седмица 4 и стойностите се запазват до 52 седмици.
Физическа активност и свързано със здравето качество на живот
В проучване при ПсА 2 и проучване при ПсА 3, пациентите, лекувани със секукинумаб 150 mg (p=0,0555 и p<0,0001) и 300 mg (p=0,0040 и p<0,0001), показват подобрение във физическата активност спрямо пациентите, лекувани с плацебо, оценено чрез Въпросник за оценка на здравето с показател за инвалидизиране (Health Assessment Questionnaire-Disability Index - HAQ-DI) съответно на седмица 24 и седмица 16. Подобрение в HAQ-DI скоровете се наблюдава независимо от предхождащата експозиция на анти-TNFα. Подобен отговор се наблюдава и в проучване при ПсА 1.
Пациентите, лекувани със секукинумаб, съобщават за значимо подобрение в свързаното със здравето качество на живот, измерено чрез Кратката форма на формуляр 36 (Обобщена оценка на здравето с физикални елементи - Health Survey Physical Component Summary - SF-36 PCS) скора (p<0,001). Също така се наблюдава статистически значимо подобрение по отношение на изследователските крайни точки, оценени чрез скалата за Функционална оценка на лечението на хронично заболяване – умора (Functional Assessment of Chronic Illness Therapy – Fatigue - FACIT-F), при 150 mg и 300 mg при сравнение с плацебо (съответно 7,97; 5,97 спрямо 1,63) и това подобрение се запазва до седмица 104 в проучване при ПсА 2.
В проучване при ПсА 1 се наблюдава подобен клиничен отговор и ефикасността се запазва до седмица 52.
Аксиален спондилоартрит (axSpA)
Анкилозиращ спондилит (AS) / Рентгенографски потвърден аксиален спондилоартрит)
Безопасността и ефикасността на секукинумаб са оценени при 816 пациенти в три рандомизирани, двойнослепи, плацебо-контролирани проучвания фаза III, при пациенти с активен анкилозиращ спондилит (АС) с Индекс за активност на заболяването анкилозиращ спондилит по Бат (Bath Ankylosing Spondylitis Disease Activity Index - BASDAI) ≥4, въпреки приложението на нестероидни противовъзпалителни средства (НСПВС), кортикостероиди или модифициращи болестта антиревматоидни средства (DMARD). Пациентите в проучване при анкилозиращ спондилит 1 (проучване при АС 1) и проучване при анкилозиращ спондилит 2 (проучване при АС 2) имат поставена диагноза АС средно от 2,7 до 5,8 години. В двете проучвания първична крайна точка е поне 20% подобрение в дефинираните от Международното дружество за оценка на спондилоартрита (Assessment of SpondyloАrthritis International Society - ASAS 20) критерии на седмица 16.
В проучването при анкилозиращ спондилит 1 (проучване при АС 1), анкилозиращ спондилит 2 (проучване при АС 2) и анкилозиращ спондилит 3 (проучване при АС 3), съответно 27,0%, 38,8% и 23,5% от пациентите са лекувани преди това с анти-TNFα средство и са преустановили лечението с анти-TNFα средството, поради липса на ефикасност или поради непоносимост (пациенти с незадоволителен отговор към анти-TNFα).
Проучване при АС 1 (MEASURE 1) оценява 371 пациенти, от които 14,8% и 33,4% провеждат съпътстващо лечение съответно с MTX или сулфасалазин. Пациентите, рандомизирани на секукинумаб, получават 10 mg/kg интравенозно на седмица 0, 2 и 4, последвани от 75 mg или 150 mg, приложени подкожно всеки месец, като се започне от седмица 8. Пациентите, рандомизирани на плацебо, при които не се наблюдава терапевтичен отговор на седмица 16 (ранна животоспасяваща терапия), и всички останали пациенти, лекувани с плацебо на седмица 24, преминават на лечение със секукинумаб (75 mg или 150 mg, приложени подкожно), последвани от същата доза всеки месец.
Проучване при АС 2 (MEASURE 2) оценява 219 пациенти, от които 11,9% и 14,2% провеждат съпътстващо лечение съответно с MTX или сулфасалазин. Пациентите, рандомизирани на секукинумаб, получават 75 mg или 150 mg, приложени подкожно на седмица 0, 1, 2, 3 и 4, последвани от същата доза всеки месец. На седмица 16, пациентите, които са рандомизирани на плацебо на изходно ниво, са рандомизирани отново да получават секукинумаб (75 mg или 150 mg, приложени подкожно) всеки месец.
Проучване при АС 3 (MEASURE 3) оценява 226 пациенти, от които 13,3% и 23,5% провеждат съпътстващо лечение съответно с MTX или сулфасалазин. Пациентите, рандомизирани на секукинумаб, получават 10 mg/kg, приложени интравенозно на седмица 0, 2 и 4, последвани от 150 mg, или 300 mg, приложени подкожно всеки месец. На седмица 16 пациентите, които са рандомизирани на плацебо на изходно ниво, са рандомизирани отново да получават секукинумаб (150 mg или 300 mg, приложени подкожно) всеки месец. Първичната крайна точка е ASAS 20 на седмица 16. Пациентите са заслепени за схемата на лечение до седмица 52, а проучването продължава до седмица 156.
Признаци и симптоми:
В проучване при АС 2 лечението със секукинумаб 150 mg води до по-голямо подобрение в показателите за активност на заболяването спрямо плацебо на седмица 16 (вж. Таблица 11).
Таблица 11 Клиничен отговор в проучване при АС 2 на седмица 16
|
Крайна точка (p-стойност спрямо плацебо) |
Плацебо (n = 74) |
75 mg (n = 73) |
150 mg (n = 72) |
|
ASAS 20 отговор, % |
28,4 |
41,1 |
61,1*** |
|
ASAS 40 отговор, % |
10,8 |
26,0 |
36,1*** |
|
hsCRP, (съотношение след BSL/BSL) |
1,13 |
0,61 |
0,55*** |
|
ASAS 5/6, % |
8,1 |
34,2 |
43,1*** |
|
ASAS частична ремисия, % |
4,1 |
15,1 |
13,9 |
|
BASDAI 50, % |
10,8 |
24,7* |
30,6** |
|
ASDAS-CRP голямо подобрение |
4,1 |
15,1* |
25,0*** |
|
* p<0,05, ** p<0,01, *** p<0,001; спрямо плацебо Всички p-стойности са коригирани за множественост на изследването въз основа на предварително определена йерархия, с изключение на BASDAI 50 и ASDAS-CRP Приписана стойност при липса на отговор при липсваща двоична крайна точка.
ASAS: Критерии на Международното дружество за оценка на спондилоартрита; BASDAI: Индекс за активност на заболяването анкилозиращ спондилит по Бат; hsCRP: високо чувствителен C-реактивен протеин; ASDAS: Скор за активност на заболяването анкилозиращ спондилит; BSL: изходно ниво |
|||
Секукинумаб 150 mg започва да действа на седмица 1 по отношение на ASAS 20 и на седмица 2 по отношение на ASAS 40 (с превъзходство спрямо плацебо) в изпитване при АС 2.
Подобрение в ASAS 20 отговора на седмица 16 се наблюдава както при пациентите, при които не е провеждана анти-TNFα терапия (68,2% спрямо 31,1%; p<0,05), така и при пациентите с незадоволителен отговор към анти-TNFα терапията (50,0% спрямо 24,1%; p<0,05) съответно при секукинумаб 150 mg спрямо плацебо.
В проучванията при АС 1 и АС 2, пациентите, лекувани със секукинумаб (150 mg в проучване при АС 2 и двете терапевтични схеми в проучване при АС 1) показват значимо подобрение на признаците и симптомите на заболяването на седмица 16, със сравнима степен на постигнат клиничен отговор и ефикасност, които се запазват до седмица 52, както при пациентите, при които не е провеждана анти-TNFα терапия, така и при пациентите с незадоволителен отговор към анти-TNFα терапията. В проучване при АС 2 от 72 пациенти, рандомизирани първоначално на секукинумаб 150 mg, 61 (84,7%) от пациентите продължават лечението на седмица 52. От 72 пациенти, рандомизирани на секукинумаб 150 mg, 45 и 35 имат съответно ASAS 20/40 отговор.
В проучването при АС 3, пациентите, лекувани със секукинумаб (150 mg и 300 mg), показват подобрение на признаците и симптомите, като имат сравним отговор за ефикасност, независимо от дозата, който превъзхожда плацебо на седмица 16 за първичната крайна точка (ASAS 20). Като цяло, честотата на отговора за ефикасност в групата с 300 mg е съответно по-висока, в сравнение с групата със 150 mg за вторичните крайни точки. По време на заслепения период, ASAS 20 и ASAS 40 отговорите са съответно 69,7% и 47,6% за 150 mg, и 74,3% и 57,4% за 300 mg на седмица 52. ASAS 20 и ASAS 40 отговорите се запазват до седмица 156 (69,5% и 47,6% за 150 mg, спрямо 74,8% и 55,6% за 300 mg). По-висока честота на отговор в полза на дозата от 300 mg се наблюдава също и в отговора за частична ремисия за ASAS (ASAS PR) на седмица 16, като стойностите се запазват до седмица 156. По-големи разлики в честотата на отговор в полза на дозата от 300 mg, в сравнение с дозата от 150 mg, се наблюдават при пациентите с незадоволителен отговор към анти-TNFα терапията (n=36), в сравнение с пациентите, при които не е провеждана анти-TNFα терапия (n=114).
Подвижност на гръбначния стълб:
Пациентите, лекувани със секукинумаб 150 mg, показват подобрение по отношение на подвижността на гръбначния стълб, оценявана чрез промяната в BASMI на седмица 16 спрямо изходното ниво и в двете проучвания при АС – проучване при АС 1 (-0,40 спрямо -0,12 за плацебо; p=0.0114) и проучване при АС 2 (-0,51 спрямо -0,22 за плацебо; p=0,0533). Това подобрение се запазва до седмица 52.
Физическа активност и свързано със здравето качество на живот:
В изпитване при АС 1 и изпитване 2, пациентите, лекувани със секукинумаб 150 mg, показват подобрение в свързаното със здравето качество на живот, измерено чрез Въпросника за качеството на живота при АС ASQoL (p=0,001) и Кратката форма на-36 Health Survey Physical Component Summary (SF 36 PCS) скора (p<0,001). Пациентите, лекувани със секукинумаб 150 mg, показват статистически значимо подобрение по отношение на изследователските крайни точки, свързани с физическата активност, оценена чрез индекса за активност на анкилозиращия спондилит по Бат (Bath Ankylosing Spondylitis Functional Index - BASFI) спрямо плацебо (-2,15 спрямо -0.68) и умората, оценена чрез скалата за Функционална оценка на лечението на хронично заболяване – умора (Functional Assessment of Chronic Illness Therapy-Fatigue - FACIT-Fatigue) спрямо плацебо (8,10 спрямо 3,30). Тези подобрения се запазват до седмица 52.
Нерентгенографски потвърден аксиален спондилоартрит (nr-axSpA)
Безопасността и ефикасността на секукинумаб са оценени при 555 пациенти в едно рандомизирано, двойносляпо, плацебо-контролирано проучване фаза III (PREVENT), състоящо се от 2-годишна основна фаза и 2-годишно продължение, при пациенти с активен нерентгенографски потвърден аксиален спондилоартрит (nr-axSpA), които изпълняват класификационните критерии на Международното дружество за оценка на спондилоартрита (Assessment of SpondyloАrthritis International Society, ASAS) за аксиален спондилоартрит (axSpA) без рентгенографски данни за промени в сакроилиачните стави, които биха отговорили на модифицираните Нюйоркски критерии за анкилозиращ спондилит (АС). Включените пациенти са имали активно заболяване, дефинирано като Индекс за активност на заболяването анкилозиращ спондилит по Бат (Bath Ankylosing Spondylitis Disease Activity Index, BASDAI) ≥4, резултат по Визуалната аналогова скала (Visual Analogue Scale, VAS) за общата болка в гърба от ≥40 (по скала от 0-100 mm), независимо от текуща или предходна терапия с нестероидини противовъзпалителни средства (НСПВС), и повишен С-реактивен протеин (CRP) и/или данни за сакроилиит от ядрено-магнитен резонанс (ЯМР). Пациентите в това проучване са имали диагноза за axSpA от средно 2,1 до 3,0 години, като 54% участниците са били от женски пол.
В проучването PREVENT 9,7% от пациентите са приемали предходна анти-TNFα терапия и са я прекратили поради липса на ефикасност или непоносимост (анти-TNFα-IR пациенти).
В проучването PREVENT 9,9% и 14,8% от пациентите са приемали съответно съпътстващо лечение с MTX или сулфасалазин. По време на двойнозаслепения период пациентите са приемали плацебо или секукинумаб в продължение на 52 седмици. Пациентите, рандомизирани за прием на секукинумаб, са получавали 150 mg подкожно на седмици 0, 1, 2, 3 и 4, след което същата доза всеки месец или инжекция със секукинумаб 150 mg веднъж месечно. Първичната крайна точка е подобрение с най-малко 40% по критериите на Международното дружество за оценка на спондилоартрита (ASAS 40) към Седмица 16 при пациентите без предходна анти-TNFα терапия.
Признаци и симптоми:
В проучването PREVENT лечението със секукинумаб 150 mg е довело до значително подобрение в показателите за активност на заболяването в сравнение с плацебо на седмица 16.
Тези показатели включват ASAS 40, ASAS 5/6, скор по BASDAI, BASDAI 50, високочувствителен CRP (hsCRP), ASAS 20 и ASAS частична ремисия в сравнение с плацебо (Таблица 1
2). Отговорите се запазват до седмица 52.
Таблица 12 Клиничен отговор в проучването PREVENT на седмица 16
|
Резултат (p-стойност спрямо плацебо) |
Плацебо |
150 mg1 |
|
Брой рандомизирани пациенти без предходна анти-TNFα терапия |
171 |
164 |
|
ASAS 40 отговор, % |
29,2 |
41,5* |
|
Общ брой рандомизирани пациенти |
186 |
185 |
|
ASAS 40 отговор, % |
28,0 |
40,0* |
|
ASAS 5/6, % |
23,7 |
40,0* |
|
BASDAI, LS средна промяна спрямо изходния скор |
-1,46 |
-2,35* |
|
BASDAI 50, % |
21,0 |
37,3* |
|
hsCRP, (съотношение след BSL/BSL) |
0,91 |
0,64* |
|
ASAS 20 отговор, % |
45,7 |
56,8* |
|
ASAS частична ремисия, % |
7,0 |
21,6* |
|
*p<0,05 спрямо плацебо Всички p-стойности са коригирани за множественост на изследването въз основа на предварително определена йерархия Приписана стойност при липса на отговор при липсваща двоична крайна точка 1секукинумаб 150 mg подкожно на седмица 0, 1, 2, 3 и 4, след което същата доза веднъж месечно
ASAS: Критерии на Международното дружество за оценка на спондилоартрита; BASDAI: Индекс за активност на заболяването анкилозиращ спондилит по Бат; hsCRP: високочувствителен C-реактивен протеин; BSL: изходно ниво; LS: метод на най-малките квадрати |
||
Секукинумаб 150 mg започва да действа още на седмица 3 по отношение на ASAS 40 при пациентите без предходна анти-TNFα терапия (превъзходство спрямо плацебо) в проучването PREVENT. Процентът пациенти, постигнали отговор ASAS 40 сред тези без преходна анти-TNFα терапия, е показан по визити на Фигура 3.
Фигура 3 ASAS 40 отговор в течение на времето при пациенти без преходна анти-TNFα терапия в проучването PREVENT до седмица 16
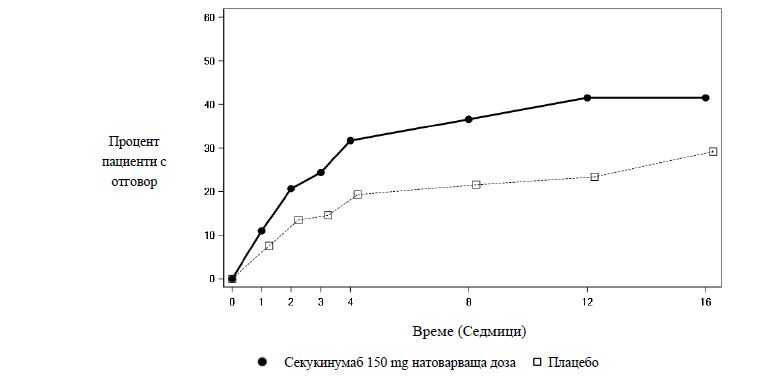
По отношение на ASAS 40 отговора също има подобрение на седмица 16 при анти-TNFα-IR пациентите, приемали секукинумаб 150 mg, в сравнение с плацебо.
Физическа активност и свързано със здравето качество на живот:
При пациентите, лекувани със секукинумаб 150 mg, е наблюдавано статистически значимо подобрение към седмица 16 във физическата активност, спрямо пациентите, лекувани с плацебо, оценено чрез BASFI (седмица 16: -1,75 спрямо -1,01, p<0,05). Пациентите, лекувани със секукинумаб съобщават за значимо подобрение в сравнение с лекуваните с плацебо към седмица 16 по отношение на свързаното със здравето качество на живот, измерено чрез ASQoL (средна промяна по метода на най-малките квадрати: седмица 16: -3,45 спрямо -1,84, p<0,05) и обобщение на физически компонент на въпросника за здравето-36 (SF-36 Physical Component Summary, SF-36 PCS) (средна промяна по метода на най-малките квадрати: седмица 16: 5,71 спрямо 2,93, p<0,05). Това подобрение се запазва до седмица 52.
Подвижност на гръбначния стълб:
Подвижността на гръбначния стълб е оценена чрез BASMI до седмица 16. По-голям брой случаи на подобрение са отчетени при пациентите, лекувани със секукинумаб, в сравнение с лекуваните с плацебо на седмици 4, 8, 12 и 16.
Намаляване на възпалението при ядрено-магнитен резонанс (ЯМР):
Признаците на възпаление са оценявани чрез ЯМР на изходното ниво и на седмица 16, и са представени като промяна спрямо изходното ниво в скора по Берлинската скала за оценка на оток в сакроилиачните стави (Berlin SI-joint oedema score) и скора ASspiMRI-a и скора по Берлинската скала за оценка на гръбначния стълб (Berlin spine score). Намаление на възпалителните симптоми в сакроилиачните стави и гръбначния стълб е наблюдавано при пациентите, лекувани със секукинумаб. Средната промяна в изходните стойности на скора по Берлинската скала за оценка на оток в сакроилиачните стави е -1,68 при пациентите, лекувани със секукинумаб 150 mg (n=180) спрямо -0,39 при пациентите, лекувани с плацебо (n=174) (p<0,05).
Педиатрична популация
Плакатен псориазис при педиатрични пациенти
Доказано е, че секукинумаб подобрява признаците и симптомите, както и качеството на живот свързано със здравето, при педиатрични пациенти на възраст на и над 6 години, които имат
Процент пациенти с отговор
Време (Седмици)
Секукинумаб 150 mg натоварваща доза Плацебо плакатен псориазис (вж. Таблица 14 и 16).
Тежък плакатен псориазис
Безопасността и ефикасността на секукинумаб са оценени в рандомизирано, двойносляпо, плацебо- и етанерцепт-контролирано проучване фаза III при педиатрични пациенти на възраст от 6 до <18 години с тежък плакатен псориазис, дефиниран чрез PASI скор ≥20, IGA mod 2011 скор 4 и BSA ангажиране ≥10%, които са кандидати за системна терапия. Приблизително 43% от пациентите са имали предходна експозиция на фототерапия, 53% на конвенционална системна терапия, 3% на биологични лекарствени продукти, а 9% са имали съпътстващ псориатичен артрит.
В педиатричното проучване 1 при псориазис са оценени 162 пациенти, които са рандомизирани да приемат ниска доза секукинумаб (75 mg за телесно тегло <50 kg или 150 mg за телесно тегло ≥50 kg), висока доза секукинумаб (75 mg за телесно тегло <25 kg, 150 mg за телесно тегло между ≥25 kg и <50 kg, или 300 mg за телесно тегло ≥50 kg) или плацебо на седмица 0, 1, 2, 3 и 4, последвано от същата доза на всеки 4 седмици, или етанерцепт. Пациентите, рандомизирани на етанерцепт, са получавали 0,8 mg/kg седмично (до максимум 50 mg). Разпределението на пациентите по телесно тегло и възраст при рандомизирането е описано в Таблица 13.
Таблица 13 Разпределение на пациентите по телесно тегло и възраст в педиатрично проучване 1 за псориазис
|
Страти при рандомизирането |
Описание |
Ниска доза секукинумаб n=40 |
Висока доза секукинумаб n=40 |
Плацебо
n=41 |
Етанерцепт
n=41 |
Общо
N=162 |
|
Възраст |
6- <12 годин и |
8 |
9 |
10 |
10 |
37 |
|
≥12- <18 годин и |
32 |
31 |
31 |
31 |
125 |
|
|
Телесно тегло |
<25 kg |
2 |
3 |
3 |
4 |
12 |
|
≥25-<50 kg |
17 |
15 |
17 |
16 |
65 |
|
|
≥50 kg |
21 |
22 |
21 |
21 |
85 |
Пациентите, рандомизирани на плацебо, при които няма отговор на седмица 12, преминават в група на ниска или висока доза секукинумаб (доза въз основа на групата по телесно тегло) и получават изпитваното лекарство на седмица 12, 13, 14 и 15, последвано от същата доза на всеки 4 седмици, като се започва от седмица 16. Първичните съвместни крайни точки са делът на пациентите, които са постигнали отговор PASI 75 и отговор IGA mod 2011 „чиста“ или „почти чиста“ кожа (0 или 1) на седмица 12.
По време на 12-седмичния плацебо-контролиран период, ефикасността на ниската и на високата доза секукинумаб са сравними по отношение на първичните съвместни крайни точки. Изчисленото съотношение на шансовете в полза и на двете дози секукинумаб е статистически значимо, както за отговора PASI 75, така и за IGA mod 2011 0 или 1.
Всички пациенти са проследявани за ефикасност и безопасност по време на 52 седмици след първата доза. Процентът пациенти, постигнали отговор PASI 75 и IGA mod 2011 „чиста“ или „почти чиста“ кожа (0 или 1), показва разлика между групите, лекувани със секукинумаб и плацебо, при първата визита на седмица 4 след изходното ниво, като разликата става по- изразена на седмица 12. Отговорът се поддържа по време на целия 52-седмичен период (вж. Таблица 14). Подобрението в степента на отговор PASI 50, 90, 100 и Индекса за дерматологично качество на живот при деца (Children’s Dermatology Life Quality Index, CDLQI) скор 0 или 1 също са запазени по време на целия 52-седмичен период.
В допълнение, честотите на отговор PASI 75, IGA 0 или 1, PASI 90 на седмици 12 и 52 и за двете групи с ниска и висока доза секукинумаб са по-високи, в сравнение с честотите на отговор за пациентите, лекувани с етанерцепт (вж. Таблица 14).
След седмица 12 ефикасността при ниската и високата доза секукинумаб са сравними, въпреки че ефикасността на по-високата доза е по-голяма при пациенти ≥50 kg. Профилите на безопасност за ниската и високата доза са сравними и съответстват с профила на безопасност при възрастни пациенти.
Таблица 14 Обобщение на клиничния отговор при педиатрични пациенти с тежък псориазис на седмица 12 и 52 (педиатрично проучване 1 при псориазис)*
|
Критерий за отговор |
Сравнение на лечението |
„тест“ |
„контрола“ |
Изчислено съотношение на шансовете |
p-стойност |
|
“тест“ спрямо „контрола“ |
n**/m (%) |
n**/m (%) |
(95% CI) |
||
|
На седмица 12*** |
|||||
|
PASI 75 |
секукинумаб ниска доза спрямо плацебо |
32/40 (80,0) |
6/41 (14,6) |
25,78 (7,08, 114,66) |
<0,0001 |
|
секукинумаб висока доза спрямо плацебо |
31/40 (77,5) |
6/41 (14,6) |
22,65 (6,31, 98,93) |
<0,0001 |
|
|
секукинумаб ниска доза спрямо етанерцепт |
32/40 (80,0) |
26/41 (63,4) |
2,25 (0,73, 7,38) |
|
|
|
секукинумаб висока доза спрямо етанерцепт |
31/40 (77,5) |
26/41 (63,4) |
1,92 (0,64, 6,07) |
|
|
|
IGA 0/1 |
секукинумаб ниска доза спрямо плацебо |
28/40 (70,0) |
2/41 (4,9) |
51,77 (10,02, 538,64) |
<0,0001 |
|
секукинумаб висока доза спрямо плацебо |
24/40 (60,0) |
2/41 (4,9) |
32,52 (6,48, 329,52) |
<0,0001 |
|
|
секукинумаб ниска доза спрямо етанерцепт |
28/40 (70,0) |
14/41 (34,1) |
4,49 (1,60, 13,42) |
|
|
|
секукинумаб висока доза спрямо етанерцепт |
24/40 (60,0) |
14/41 (34,1) |
2,86 (1,05, 8,13) |
|
|
|
PASI 90 |
секукинумаб ниска доза спрямо плацебо |
29/40 (72,5) |
1/41 (2,4) |
133,67 (16,83, 6395,22) |
<0,0001 |
|
секукинумаб висока доза спрямо плацебо |
27/40 (67,5) |
1/41 (2,4) |
102,86 (13,22, 4850,13) |
<0,0001 |
|
|
секукинумаб ниска доза спрямо етанерцепт |
29/40 (72,5) |
12/41 (29,3) |
7,03 (2,34, 23,19) |
|
|
|
секукинумаб висока доза спрямо етанерцепт |
27/40 (67,5) |
12/41 (29,3) |
5,32 (1,82, 16,75) |
|
|
|
На седмица 52 |
|||||
|
PASI 75 |
секукинумаб ниска доза спрямо етанерцепт |
35/40 (87,5) |
28/41 (68,3) |
3,12 (0,91, 12,52) |
|
|
секукинумаб висока доза спрямо етанерцепт |
35/40 (87,5) |
28/41 (68,3) |
3,09 (0,90, 12,39) |
||
|
IGA 0/1 |
секукинумаб ниска доза спрямо етанерцепт |
29/40 (72,5) |
23/41 (56,1) |
2,02 (0,73, 5,77) |
|
|
секукинумаб висока доза спрямо етанерцепт |
30/40 (75,0) |
23/41 (56,1) |
2,26 (0,81, 6,62) |
||
|
PASI 90 |
секукинумаб ниска доза спрямо етанерцепт |
30/40 (75,0) |
21/41 (51,2) |
2,85 (1,02, 8,38) |
|
|
секукинумаб висока доза спрямо етанерцепт |
32/40 (80,0) |
21/41 (51,2) |
3,69 (1,27, 11,61) |
||
|
* приписана стойност при липса на отговор е използвана за обработка на липсващите стойности ** n е броят на отговорилите, m = брой оценени пациенти *** удължен прозорец за визита на седмица 12 Съотношението на шансовете, 95% доверителен интервал и p-стойностите са от точен логистичен регресионен модел с група на лечение, категория по телесно тегло на изходно ниво и категория по възраст като фактори |
|||||
По-голям процент педиатрични пациенти, лекувани със секукинумаб, съобщават за подобрение на качеството на живот, свързано със здравето, оценено чрез CDLQI скор 0 или 1, в сравнение с плацебо на седмица 12 (ниска доза 44,7%, висока доза 50%, плацебо 15%). С времето, до седмица 52 включително, и двете дозови групи на секукинумаб са с числено по-високи резултати, спрямо групата на етанерцепт (ниска доза 60,6%, висока доза 66,7%, етанерцепт 44,4%).
Умерен до тежък плакатен псориазис
Прогнозира се, че секукинумаб ще е ефективен за лечение на педиатрични пациенти с умерен плакатен псориазис въз основа на доказаната връзка между ефикасността и отговора към експозицията при възрастни пациенти с умерен до тежък плакатен псориазис, както и на сходствата в протичането на заболяването, патофизиологията и лекарствения ефект при възрастни и педиатрични пациенти при еднакви нива на експозиция.
Освен това безопасността и ефикасността на секукинумаб са оценени в многоцентрово открито проучване фаза III, с две рамена, с паралелни групи при педиатрични пациенти на възраст от 6 до <18 години, с умерен до тежък плакатен псориазис, дефиниран чрез PASI скор ≥12, IGA mod 2011 скор ≥3 и BSA ангажиране ≥10%, които са кандидати за системна терапия.
В педиатрично проучване 2 при псориазис са оценени 84 пациенти, които са рандомизирани на ниска доза секукинумаб (75 mg за телесно тегло <50 kg или 150 mg за телесно тегло ≥50 kg) или висока доза секукинумаб (75 mg за телесно тегло <25 kg, 150 mg за телесно тегло между ≥25 kg и <50 kg или 300 mg за телесно тегло ≥50 kg) на седмица 0, 1, 2, 3 и 4, последвано от същата доза на всеки 4 седмици. Разпределението на пациентите по телесно тегло и възраст при рандомизирането е описано в Таблица 15.
|
Подгрупи |
Описание |
Секукинумаб ниска доза n=42 |
Секукинумаб висока доза n=42 |
Общо
N=84 |
|
Възраст |
6-<12 години |
17 |
16 |
33 |
|
≥12-<18 години |
25 |
26 |
51 |
|
|
Телесно тегло |
<25 kg |
4 |
4 |
8 |
|
≥25-<50 kg |
13 |
12 |
25 |
|
|
≥50 kg |
25 |
26 |
51 |
PASI 75 и отговор IGA mod 2011 „чиста“ или „почти чиста“ кожа (0 или 1) на седмица 12.
Ефикасността на ниската и високата доза на секукинумаб са сравними и показват статистически значимо подобрение, в сравнение с историческите данни за плацебо, за първичните съвместни крайни точки. Изчислената постериорна вероятност за позитивен ефект от лечението е 100%.
Пациенти са проследени за ефикасност в рамките на период от 52 седмици след първото приложение. Ефикасност (дефиниране чрез PASI 75 и IGA mod 2011“чиста“ или „почти чиста“ кожа [0 или 1]) е наблюдавана още след първата визита на седмица 2 след изходното ниво, а процентът пациенти, които са постигнали отговор PASI 75 и IGA mod 2011“чиста“ или „почти чиста“ кожа (0 или 1) се повишава до седмица 24 и се запазва до седмица 52. Подобрение в PASI 90 и PASI 100 също се наблюдава на седмица 12, повишава се до седмица 24 и се запазва до седмица 52 (вж. Таблица 16).
Профилите на безопасност на ниската доза и на високата доза са сравними и съответстват на профила на безопасност при възрастни пациенти.
Таблица 16 Обобщение на клиничния отговор при умерен до тежък плакатен псориазис при педиатрични пациенти на седмици 12 и 52 (педиатрично проучване 2 при псориазис)*
|
|
Седмица 12 |
Седмица 52 |
||
|
Секукинумаб ниска доза |
Секукинумаб висока доза |
Секукинумаб ниска доза |
Секукинумаб висока доза |
|
|
Брой пациенти |
42 |
42 |
42 |
42 |
|
Отговор PASI 75 n (%) |
39 (92,9%) |
39 (92,9%) |
37 (88,1%) |
38 (90,5%) |
|
Отговор IGA mod 2011 “чиста“ или „почти чиста“ кожа n (%) |
33 (78,6%) |
35 (83,3%) |
36 (85,7%) |
35 (83,3%) |
|
Отговор PASI 90 n (%) |
29 (69%) |
32 (76,2%) |
32 (76,2%) |
35 (83,3%) |
|
Отговор PASI 100 n (%) |
25 (59,5%) |
23 (54,8%) |
22 (52,4%) |
29 (69,0%) |
|
* приписана стойност при липса на отговор е използвана за обработка на липсващите стойности |
||||
Тези резултати в популацията от педиатрични пациенти с умерен до тежък плакатен псориазис потвърждават прогнозните предположения въз основа на връзката между ефикасността и отговора към експозицията при възрастни пациенти, споменати по-горе.
В групата с ниска доза 50% и 70,7% от пациентите са постигнали CDLQI скор 0 или 1, съответно на седмица 12 и 52. В групата с висока доза 61,9% и 70,3% са постигнали CDLQI скор 0 или 1, съответно на седмица 12 и 52.
Ювенилен идиопатичен артрит (ЮИА)
Артрит, свързан с ентезит (ERA) и ювенилен псориатичен артрит (ЮПсА)
Ефикасността и безопасността на секукинумаб са оценени при 86 пациенти в едно двойносляпо, плацебо-контролирано, оценяващо настъпили събития, рандомизирано, фаза III проучване от три части при пациенти на възраст 2 до <18 години с активен ERA или ЮПсА, диагностицирани въз основа на модифицирани критерии на Международна лига на асоциациите за ревматология (International League of Associations for Rheumatology, ILAR) за класифициране на ЮИА. Проучването се състои от открита част (Част 1), в която всички пациенти получават секукинумаб до седмица 12. Пациенти, показващи ЮИА ACR 30 отговор на седмица 12, са включени в двойносляпата фаза Част 2, и са рандомизирани 1:1 да продължат лечението със секукинумаб или да започнат лечение с плацебо (рандомизирано оттегляне) до седмица 104 или докато настъпи обостряне. Пациенти с настъпило обостряне, след това са включени в открито лечение със секукинумаб до седмица 104 (Част 3).
Подтиповете пациенти с ЮИА при включване в проучването са: 60,5% с ERA и 39,5% с ЮПсА, които са имали недостатъчен отговор, или са с непоносимост към ≥1 модифициращо болестта антиревматоидно средство (DMARD) и ≥1 нестероидно противовъзпалително средство (НСПВС). На изходно ниво употреба на MTX се съобщава при 65,1% от пациентите; (63,5% [33/52] от пациентите с ERA и 67,6% [23/34] от пациентите с ЮПсА). При 12 от 52 пациенти с ERA е прилагано съпътстващо лечение със сулфасалазин (23,1%). При пациенти с телесно тегло на изходно ниво <50 kg (n=30) е прилагана доза 75 mg, а при пациенти с телесно тегло ≥50 kg (n=56) е прилагана доза 150 mg. Възрастта на изходно ниво е в границите от 2 до 17 години, като 3 пациенти са на възраст между 2 до <6 години, 22 пациенти са на възраст 6 до <12 години и 61 пациенти са на възраст 12 до <18 години. На изходно ниво скорът за активност на заболяването при ювенилен артрит (Juvenile Arthritis Disease Activity Score, JADAS-27) е 15,1 (SD:7,1).
Първичната крайна точка е времето до обостряне в периода на рандомизираното оттегляне (Част 2). Обострянето на заболяването е дефинирано като ≥30% влошаване в поне три от шестте критерия за ЮИА ACR отговор и ≥30% подобрение в не повече от един от шестте критерия за ЮИА ACR отговор и минимум две активни стави.
В края на Част 1, 75 от 86 (87,2%) пациенти показват ЮИА ACR 30 отговор и са включени в Част 2.
Проучването постига първичната си крайна точка като показва статистически значимо удължаване на времето до обостряне на заболяването при пациенти, лекувани със секукинумаб, спрямо плацебо в Част 2. Рискът от обостряне е намален със 72% при пациенти, лекувани със секукинумаб, спрямо пациенти, лекувани с плацебо в Част 2 (коефициент на риск = 0,28, 95% CI: 0,13 до 0,63, p<0,001) (Фигура 4 и Таблица 17). По време на Част 2 общо 21 пациенти в групата с плацебо са получили събитие с обостряне (11 с ЮПсА и 10 с ERA) спрямо 10 пациенти в групата със секукинумаб (4 с ЮПсА и 6 с ERA).
Фигура 4 Оценки по Kaplan-Meier за времето до възникване на обостряне в Част 2
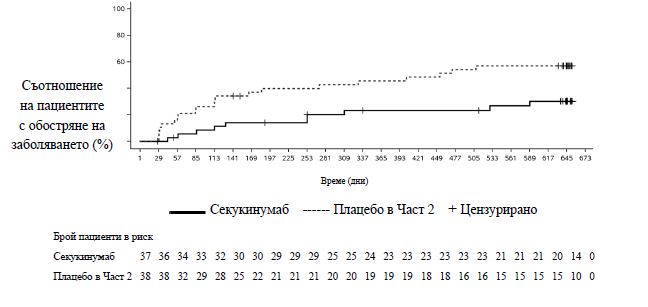
Таблица 17 Анализ на преживяемостта за времето до обостряне на заболяването – Част 2
|
|
Секукинумаб (N=37) |
Плацебо в Част 2 (N=38) |
|
Брой събития с обостряне в края на Част 2, n (%) |
10 (27,0) |
21 (55,3) |
|
Оценки по Kaplan-Meier: |
|
|
|
Медиана, в дни (95% CI) |
NC (NC, NC) |
453,0 (114.0, NC) |
|
Процент без обостряне на 6 месеца (95% CI) |
85,8 (69,2, 93,8) |
60,1 (42,7, 73,7) |
|
Процент без обостряне на 12 месеца (95% CI) |
76,7 (58,7, 87,6) |
54,3 (37,1, 68,7) |
|
Процент без обостряне на 18 месеца (95% CI) |
73,2 (54,6, 85,1) |
42,9 (26,7, 58,1) |
|
Коефициент на риск спрямо плацебо: оценка (95% CI) |
0,28 (0,13, 0,63) |
|
|
Стратифициран Log-rank тест, p- стойност |
<0,001** |
|
|
Анализът е проведен за всички рандомизирани пациенти, които са получили поне една доза от проучваното лекарство в Част 2. Секукинумаб: всички пациенти, които не са приемали плацебо. Плацебо в Част 2: всички пациенти, които са приемали плацебо в Част 2 и секукинумаб в друг(и) период(и). NC = не може да се изчисли. ** = Статистически значимо при едностранно ниво на значимост 0,025. |
||
В откритата Част 1 всички пациенти са получавали секукинумаб до седмица 12. На седмица 12 83,7%, 67,4% и 38,4% от децата са съответно с ЮИА ACR 50, 70 и 90 отговор (Фигура 5). Началото на действието на секукинумаб настъпва още на седмица 1. На седмица 12 скорът JADAS-27 е 4,64 (SD:4,73), а средното понижение от изходно ниво в JADAS-27 е -10,487 (SD:7,23).
Фигура 5 ЮИА ACR 30/50/70/90 отговор при участници до седмица 12 в Част 1*
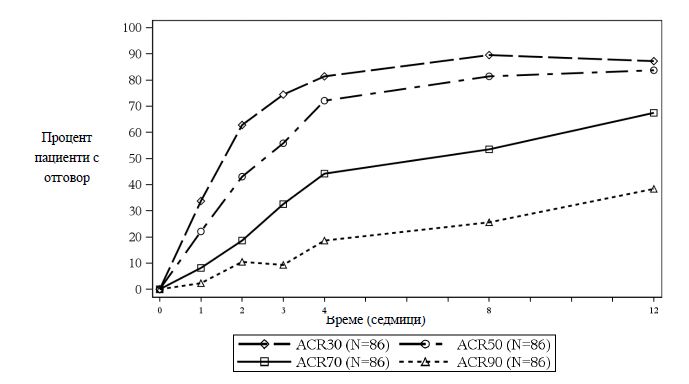
*за обработката на липсващи стойности е използвана импутация на пациенти без отговор
Данните във възрастовата група от 2 до <6 години са неубедителни поради малкия брой включени в проучването пациенти на възраст под 6 години.
Европейската агенция по лекарствата освобождава от задължението за предоставяне на резултатите от проучванията с Cosentyx при плакатен псориазис при педиатрични пациенти на възраст от раждането до под 6 години и при хроничен идиопатичен артрит при педиатрични пациенти на възраст от раждането до под 2 години (вж. точка 4.2 за информация относно употреба в педиатрията).
5.2 Фармакокинетични свойства
Повечето фармакокинетични свойства, наблюдавани при пациенти с плакатен псориазис, псориатичен артрит и анкилозиращ спондилит са сходни.
Абсорбция
След подкожно приложение на единична доза от 300 mg, под формата на разтвор, при здрави доброволци, секукинумаб достига пикова концентрация в серума 43,2±10,4 μg/ml между 2 и 14 дни след прилагане на дозата.
Въз основа на данни от популационен фармакокинетичен анализ, след подкожно приложение на единична доза от 150 mg или 300 mg при пациенти с плакатен псориазис, секукинумаб достига пикова концентрация в серума съответно 13,7±4,8 μg/ml или 27,3±9,5 μg/ml между 5 и 6 дни след прилагане на дозата.
След първоначалното ежеседмично приложение през първия месец, времето за достигане на максимална концентрация е между 31 и 34 дни, въз основа на данни от популационния фармакокинетичен анализ.
Въз основа на симулационни данни, пиковите концентрации в стационарно състояние (Cmax,ss) след подкожно приложение на 150 mg и 300 mg са били съответно 27,6 μg/ml и 55,2 μg/ml.
Данните от популационния фармакокинетичен анализ показват, че стационарно състояние се достига след 20 седмици при ежемесечно приложение.
Сравнено с експозицията след прилагане на единична доза, данните от популационния фармакокинетичен анализ показват, че пациентите имат 2-кратно повишение на пиковата серумна концентрация и площта под кривата (AUC) след многократно ежемесечно приложение, в периода на поддържащата терапия.
Популационният фармакокинетичен анализ показва, че секукинумаб се абсорбира със средна абсолютна бионаличност 73% при пациентите с плакатен псориазис. В проучванията абсолютната бионаличност варира между 60 и 77%, където е изчислявана.
Бионаличността на секукинумаб при пациенти с ПсА е 85% въз основа на популационен фармакокинетичен модел.
След подкожно приложение на една инжекция от 300 mg инжекционен разтвор в предварително напълнена спринцовка при пациенти с плакатен псоризис, системната експозиция на секукинумаб е подобна на предходно наблюдаваната при две инжекции от 150 mg.
Разпределение
Средният обем на разпределение в крайната фаза (Vz), след еднократно интравенозно приложение варира от 7,10 до 8,60 литра при пациентите с плакатен псориазис, което предполага, че разпределението на секукинумаб в периферните компартименти е ограничено.
Биотрансформация
По-голямата част от елиминирането на IgG се осъществява чрез вътреклетъчен катаболизъм, след течнофазова или рецептор-медиирана ендоцитоза.
Елиминиране
Средният системен клирънс (CL) след еднократно интравенозно приложение при пациенти с плакатен псориазис варира от 0,13 до 0,36 литра/ден. При популационен фармакокинетичен анализ средният системен клирънс (CL) е 0,19 литра/ден при пациенти с плакатен псориазис. CL не се повлиява от пола. Клирънсът е дозо- и време-независим.
Средният елиминационен полуживот, изчислен при популационния фармакокинетичен анализ, е 27 дни при пациенти с плакатен псориазис, варирайки от 18 до 46 дни в различните проучвания при псориазис с интравенозно приложение.
Линейност/нелинейност
Фармакокинетиката на секукинумаб след еднократно и многократно приложение при пациенти с плакатен псориазис е определена в няколко проучвания с интравенозно приложение на дози от 1 x 0,3 mg/kg до 3 x 10 mg/kg и подкожно приложение на дози от 1 x 25 mg до многократно приложение на дози от 300 mg. Експозицията е пропорционална на дозата при всички схеми на приложение.
Специални популации
Пациенти в старческа възраст
Въз основа на популационния фармакокинетичен анализ, в който участват ограничен брой пациенти в старческа възраст (n=71 за възраст ≥65 години и n=7 за възраст ≥75 години), клирънсът при пациентите в старческа възраст и пациентите на възраст под 65 години е бил подобен.
Пациенти с бъбречно или чернодробно увреждане
Липсват фармакокинетични данни при пациенти с бъбречно или чернодробно увреждане. Бъбречното елиминиране на интактния секукинумаб - IgG моноклонално антитяло, се очаква да е ниско и от минимално значение. IgG се елиминира предимно чрез катаболизъм и увреждането на черния дроб не се очаква да има влияние върху клирънса на секукинумаб.
Влияние на теглото върху фармакокинетиката
Клирънсът и обемът на разпределение на секукинумаб се повишават при повишаване на телесното тегло.
Педиатрична популация
Плакатен псориазис
В сборни данни от двете педиатрични проучвания, на пациентите с умерен до тежък плакатен псориазис (на възраст от 6 до под 18 години) е прилаган секукинумаб при препоръчителната схема на прилагане за педиатрични пациенти. На седмица 24 пациентите с телесно тегло ≥25 и <50 kg са имали средна ± SD най-ниска концентрация в стационарно състояние 19,8 ± 6,96 μg/ml (n=24) след 75 mg секукинумаб, а пациентите с телесно тегло ≥50 kg са имали средна ± SD най-ниска концентрация 27,3 ± 10,1 μg/ml (n=36) след 150 mg секукинумаб. Средната ± SD най-ниска концентрация в стационарно състояние при пациенти с телесно тегло <25 kg (n=8) е била 32,6 ± 10,8 μg/ml на седмица 24 след доза 75 mg.
Ювенилен идиопатичен артрит
В едно педиатрично проучване при пациентите с ERA и ЮПсА (на възраст 2 до по-малко от 18 години) е прилаган секукинумаб в препоръчителния педиатричен дозов режим. На седмица 24 пациентите с тегло <50 kg и с тегло ≥50 kg имат средна ± SD най-ниска концентрация в стационарно състояние 25,2±5,45 μg/ml (n=10) и съответно 27,9±9,57 μg/ml (n=19).
5.3 Предклинични данни за безопасност
Неклиничните данни не показват особен риск за хора (възрастни или педиатрични пациенти) на базата на конвенционалните фармакологични проучвания за безопасност, проучвания за токсичност при многократно прилагане и репродуктивна токсичност, или кръстосана тъканна реактивност.
Не са провеждани проучвания при животни, за да се оцени канцерогенния потенциал на секукинумаб.
6. ФАРМАЦЕВТИЧНИ ДАННИ
6.1 Списък на помощните вещества
Трехалоза дихидрат
Хистидин
Хистидинов хидрохлорид монохидрат
Метионин
Полисорбат 80
Вода за инжекции
6.2 Несъвместимости
При липса на проучвания за несъвместимости този лекарствен продукт не трябва да се смесва с други лекарствени продукти.
6.3 Срок на годност
2 години
Ако е необходимо, Cosentyx може да се съхранява извън хладилник за еднократен период до 4 дни на стайна температура, която не надвишава 30°C.
6.4 Специални условия на съхранение
Да се съхранява в хладилник (2°C - 8°C). Да не се замразява.
Съхранявайте в оригиналната опаковка, за да се предпази от светлина.
6.5 Вид и съдържание на опаковката
Cosentyx 150 mg инжекционен разтвор в предварително напълнена спринцовка
Cosentyx 150 mg инжекционен разтвор в предварително напълнена спринцовка се доставя в 1 ml предварително напълнена стъклена спринцовка с глава на буталото от бромобутилова гума със силиконово покритие, с прикрепена игла 27G x ½″ и с твърда защитна капачка на иглата от стирен-бутадиенова гума, поставена в автоматичен предпазител на иглата от поликарбонат.
Cosentyx 150 mg инжекционен разтвор в предварително напълнена спринцовка се предлага в единични опаковки, съдържащи 1 или 2 предварително напълнени спринцовки и в групови опаковки, съдържащи 6 (3 опаковки по 2) предварително напълнени спринцовки.
Cosentyx 300 mg инжекционен разтвор в предварително напълнена спринцовка
Cosentyx 300 mg инжекционен разтвор в предварително напълнена спринцовка се доставя в 2,25 ml предварително напълнена стъклена спринцовка с глава на буталото от бромобутилова гума със силиконово покритие, с прикрепена игла 27G x ½″ и с твърда защитна капачка на иглата от синтетична полиизопренова гума, поставена в автоматичен предпазител на иглата от поликарбонат.
Cosentyx 300 mg инжекционен разтвор в предварително напълнена спринцовка се предлага в единични опаковки, съдържащи 1 предварително напълнена спринцовка и в групови опаковки, съдържащи 3 (3 опаковки по 1) предварително напълнени спринцовки.
Cosentyx 150 mg инжекционен разтвор в предварително напълнена писалка
Cosentyx 150 mg инжекционен разтвор в предварително напълнена писалка се доставя в предварително напълнена спринцовка за еднократна употреба, поставена в триъгълна писалка с прозрачно прозорче и етикет. Предварително напълнената спринцовка в писалката представлява 1 ml стъклена спринцовка с глава на буталото от бромобутилова гума със силиконово покритие, с прикрепена игла 27G x ½″ и с твърда защитна капачка на иглата от стирен-бутадиенова гума.
Cosentyx 150 mg инжекционен разтвор в предварително напълнена писалка се предлага в единични опаковки, съдържащи 1 или 2 предварително напълнени писалки и в групови опаковки, съдържащи 6 (3 опаковки по 2) предварително напълнени писалки.
Cosentyx 300 mg инжекционен разтвор в предварително напълнена писалка
Cosentyx 300 mg инжекционен разтвор в предварително напълнена писалка се доставя в предварително напълнена спринцовка за еднократна употреба, поставена в квадратна писалка с прозрачно прозорче и етикет. Предварително напълнената спринцовка в писалката представлява 2,25 ml стъклена спринцовка с глава на буталото от бромобутилова гума със силиконово покритие, с прикрепена игла 27G x ½″ и с твърда защитна капачка на иглата от синтетична полиизопренова гума.
Cosentyx 300 mg инжекционен разтвор в предварително напълнена писалка се предлага в единични опаковки, съдържащи 1 предварително напълнена писалка и в групови опаковки, съдържащи 3 (3 опаковки по 1) предварително напълнени писалки.
Не всички видове опаковки могат да бъдат пуснати в продажба.
6.6 Специални предпазни мерки при изхвърляне и работа
Cosentyx 150 mg инжекционен разтвор в предварително напълнена спринцовка
Cosentyx 150 mg инжекционен разтвор се предоставя в предварително напълнена спринцовка за еднократна употреба за индивидуално приложение. Спринцовката трябва да се извади от хладилника 20 минути преди инжектирането, за да може да достигне стайна температура.
Cosentyx 300 mg инжекционен разтвор в предварително напълнена спринцовка
Cosentyx 300 mg инжекционен разтвор се предоставя в предварително напълнена спринцовка за еднократна употреба за индивидуално приложение. Спринцовката трябва да се извади от хладилника 30-45 минути преди инжектирането, за да може да достигне стайна температура.
Cosentyx 150 mg инжекционен разтвор в предварително напълнена писалка
Cosentyx 150 mg инжекционен разтвор се предоставя в предварително напълнена писалка за еднократна употреба за индивидуално приложение. Писалката трябва да се извади от хладилника 20 минути преди инжектирането, за да може да достигне стайна температура.
Cosentyx 300 mg инжекционен разтвор в предварително напълнена писалка
Cosentyx 300 mg инжекционен разтвор се предоставя в предварително напълнена писалка за еднократна употреба за индивидуално приложение. Писалката трябва да се извади от хладилника 30-45 минути преди инжектирането, за да може да достигне стайна температура
Преди употреба се препоръчва предварително напълнената спринцовка или предварително напълнената писалка да се огледа добре. Течността трябва да бъде бистра. На цвят може да варира от безцветна до бледожълта. Възможно е да видите малко мехурче въздух, което е нормално. Не използвайте, ако течността съдържа лесно видими частици, ако е мътна или видимо кафява. Подробни указания относно начина на употреба са предоставени в листовката.
Неизползваният лекарствен продукт или отпадъчните материали от него трябва да се изхвърлят в съответствие с местните изисквания.
7. ПРИТЕЖАТЕЛ НА РАЗРЕШЕНИЕТО ЗА УПОТРЕБА
Novartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublin 4
Ирландия
8. НОМЕР(А) НА РАЗРЕШЕНИЕТО ЗА УПОТРЕБА
Cosentyx 150 mg инжекционен разтвор в предварително напълнена спринцовка
EU/1/14/980/002
EU/1/14/980/003
EU/1/14/980/006
Cosentyx 300 mg инжекционен разтвор в предварително напълнена спринцовка
EU/1/14/980/008-009
Cosentyx 150 mg инжекционен разтвор в предварително напълнена писалка
EU/1/14/980/004
EU/1/14/980/005
EU/1/14/980/007
Cosentyx 300 mg инжекционен разтвор в предварително напълнена писалка
EU/1/14/980/010-011
9. ДАТА НА ПЪРВО РАЗРЕШАВАНЕ/ПОДНОВЯВАНЕ НА РАЗРЕШЕНИЕТО ЗА УПОТРЕБА
Дата на първо разрешаване: 15 януари 2015 г.
Дата на последно подновяване: 03 септември 2019 г.
10. ДАТА НА АКТУАЛИЗИРАНЕ НА ТЕКСТА
19 януари 2023 г.
Подробна информация за този лекарствен продукт е предоставена на уебсайта на Европейската агенция по лекарствата http://www.ema.europa.eu [3].